


INTRODUCTION
Cystic fibrosis (CF) is a complex genetic disorder resulting from a malfunction in chloride channel expression, particularly affecting the CF transmembrane conductance regulator (CFTR protein) and inherited in an autosomal recessive pattern1.
In recent years, advancements in molecular genetics have revolutionized our understanding of CF, enabling the identification and characterization of numerous CFTR gene variants linked to the disease2. Some mutations entirely disrupt the gene’s normal function, while others may only have a partial effect3. Reporting newly identified variants adds further value to comprehending the genetic foundation of the disease and its clinical manifestations.
CASE REPORT
We present a case of a male patient wherein the variant c.4345_*486delinsTTG emerges as a novel addition to the CF mutation spectrum. At three months of age, a 21-year-old male of Andalusian descent presented with symptoms suggestive of CF following hospitalization for hypotonic dehydration. While the patient’s paternal grandfather had a personal history of chronic bronchitis, the father has a history of gastric ulcer, and the mother has a history of duodenal ulcer; no other significant medical conditions were reported in the family. The initial genetic panel analysis revealed carrier status for the maternally inherited F508del mutation (Figure 1).
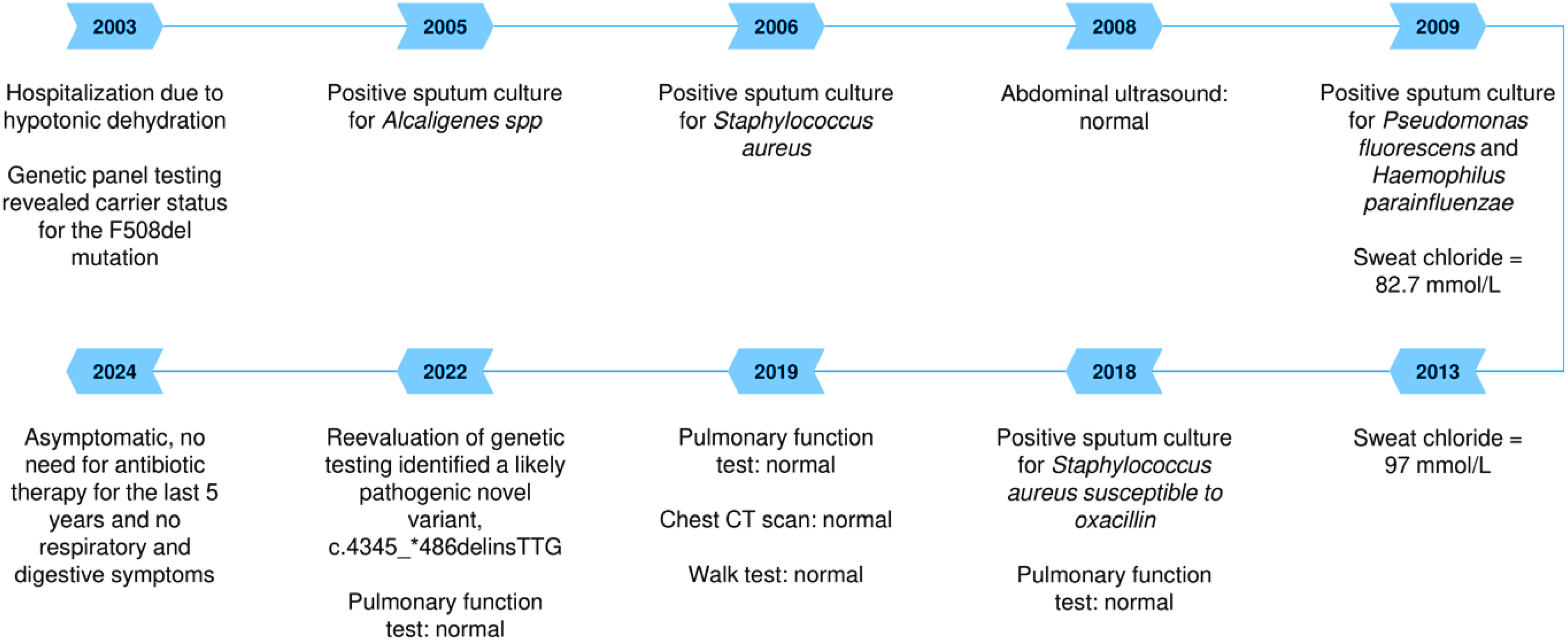
However, the patient exhibited elevated sweat chloride levels in two independent determinations (82.7 and 97 mmol/L; reference values for a positive test: 60 mmol/L)2. Over time, the patient has had positive sputum cultures for various pathogens: Alcaligenes spp. in 2005, Staphylococcus aureus in 2006, Pseudomonas fluorescens and Hemophilus parainfluenzae in 2009, and S. aureus susceptible to oxacillin in 2018, requiring intermittent antibiotic therapy, although specific details regarding the treatment regimens are unavailable.
The results of pulmonary function tests conducted in the years 2022, 2019 and 2018 revealed forced vital capacity (FVC) measurements of 124%, 111.5%, and 125.8%, respectively. Correspondingly, forced expiratory volume in one second (FEV1) values were 130%, 121.9%, and 134.1%, while the FEV1/FVC ratios were 87%, 90.26%, and 88.02%, respectively. A walk test and high-definition chest CT scan showed minimal desaturation at 99%, covering 648 m and revealing no significant pulmonary abnormalities, respectively, without identifying bronchiectasis. Additionally, the abdominal ultrasound showed normal findings, and there were no clinical symptoms suggestive of gastrointestinal problems or pancreatic insufficiency.
In 2022, a re-evaluation of genetic testing using alternative techniques was conducted. First, an assessment of gene dosage of all coding exons of the CFTR gene (SALSA MLPA Probemix P091-D2) was conducted through capillary electrophoresis of MLPA products (AB3500 Genetic Analyzer from Applied Biosystem). This study yielded inconclusive results, attributed to the non-hybridization of one of the two probes in the exon 27 region.
Subsequently, exome sequencing was performed through capture and enrichment of the exonic regions and flanking intronic regions of the genes (SureSelect custom Agilent Constitutional Panel 17 Mb), followed by massive sequencing using NextSeqTM (Illumina), specifically targeting the CFTR gene. The results returned negative findings but showed a sharp drop in coverage starting from the midpoint of exon 27.
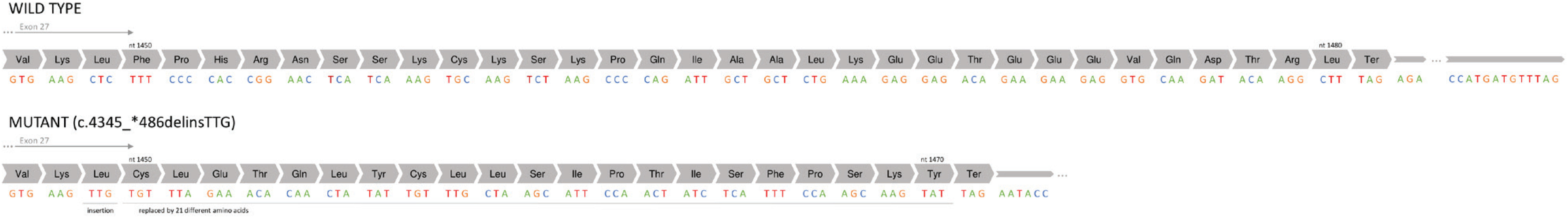
Finally, definitive diagnosis was achieved through PCR amplification targeting the exon 27 region and non-coding 3’UTR region of the CFTR gene, and Sanger sequencing of the amplification products (AB3500 Genetic Analyzer from Applied Biosystem). The analysis was conducted on the index case and his parents. This analysis identified a likely pathogenic novel c.4345_*486delinsTTG; p.(Phe1450_Leu1480delinsCysLeuGluThrGlnLeuTyrCysLeuLeuSerIle-ProThrIleSerPheProSerLysTyr) variant, confirmed to be of paternal origin, thereby confirming the compound heterozygosity of the variant with the F508del mutation of maternal origin and the diagnosis of CF. Variant classification was based on the recommendations of the American College of Medical Genetics and Genomics4. This compound heterozygous genotype illustrates the importance of comprehensive genetic testing in diagnosing CF accurately, especially in cases where conventional testing methods may miss fewer common variants.
Currently, the patient is not undergoing any specific treatment for CF, including bronchodilators or other maintenance therapies. He has not experienced pulmonary exacerbations or required hospitalizations. Despite the confirmed diagnosis, the patient has demonstrated reluctance to initiate treatment, citing a perceived absence of significant symptoms and overall good health. This reflects the mild phenotype associated with his condition and highlights the importance of individualized, patient-centered management.
DISCUSSION
The c.4345_*486delinsTTG variant represents a complex genetic alteration characterized by the deletion of 99 nucleotides within the coding region and 486 nucleotides within the non-coding 3’UTR region. This molecular rearrangement results in the deletion of 31 amino acids from positions 1450 to 1480 and the production of a protein with 21 distinct amino acids (CysLeuGluThrGlnLeuTyrCysLeuLeuSerIle-ProThrIleSerPheProSerLysTyr) due to the insertion of TTG (Figure 2), suggesting potential alterations in mRNA stability and protein structure. Further studies on mRNA are necessary to fully understand the impact of these alterations on gene expression and protein function.
Despite its intricate nature, the c.4345_*486delinsTTG variant remains notably absent from widely referenced databases such as gnomAD, ClinVar, HGMD, and CFTR2. As of the most recent file (25 September 2024), a total of 1167 variants are annotated on the CFTR2 website5 Among these, 1085 are classified as CF-causing, 55 are variants of varying clinical consequence, and 27 are non-CF-causing. Since approximately 2500 mutations have been identified in the CFTR gene, this indicates that 53% of mutations are still of undetermined clinical significance. Moreover, the lack of documentation in scientific literature or reports from CF-affected individuals further accentuates the novelty of this variant.
Its detection in this patient, who presented with elevated sweat chloride levels and positive sputum cultures for various pathogens, suggests an association with CF-related clinical manifestations. However, the absence of significant pulmonary pathology, as evidenced by consistently normal or above-normal pulmonary function test results, a normal walk test, and high-definition chest CT scans without bronchiectasis, highlights the mild phenotype of the disease. These findings, along with the patient’s lack of gastrointestinal symptoms or pancreatic insufficiency, reinforced the decision to adopt a conservative management approach. This approach aligned with the patient’s reluctance to pursue treatment and his perception of being in good health.
Diagnosing CF in adulthood can be challenging, particularly in patients presenting with mild or atypical phenotypes6,7. This case highlights the importance of considering a wide spectrum of disease presentations, which may range from classical forms with severe pulmonary and digestive involvement to mild phenotypes with limited clinical manifestations. Although CFTR modulators were not deemed necessary for this patient due to his mild presentation, their use could be considered in similar cases to enhance residual CFTR function8,9. This emphasizes the need for personalized therapeutic approaches based on the genetic and clinical profile of each patient.
CONCLUSION
The identification of the novel variant c.4345_*486delinsTTG underscores the necessity for more comprehensive genetic diagnostic approaches beyond conventional methodologies in cases where two causing CFTR variants, in trans disposition, are not identified.
This variant represents a significant finding within the context of CF genetics, particularly due to its association with a mild phenotype of the disease. The patient’s stable clinical condition further emphasizes the need for tailored management strategies that integrate advanced genetic diagnostics and thorough clinical evaluations.
Future research should focus on assessing the functional and clinical impact of rare CFTR variants, particularly their effects on mRNA stability and protein function. Additionally, investigating the potential therapeutic role of CFTR modulators in mild phenotypes could provide new insights into managing patients with residual CFTR function. These findings underline the importance of continuously advancing genetic and clinical strategies to improve outcomes for patients with rare CFTR variants.