



INTRODUCTION
Chronic human immunodeficiency virus (HIV) infection weakens the immune system and makes people vulnerable to a variety of opportunistic infections that are not found in immunocompetent hosts. Most people who were diagnosed with HIV in the mid 1980s died quickly due to the lack of specific medications to reduce virus replication. However, the introduction of highly active antiretroviral therapy (HAART), which is also known as antiretroviral therapy or ART, nearly 20 years ago, converted HIV into a chronic disorder with a much better prognosis, and people on ART are now living with the disease for more than three decades1.
While opportunistic infections continue to be a major issue in HIV-infected people, an outstanding proportion of data ranging from epidemiological investigations to cell-culture models, underscores the significance of noninfectious disorders in this community as well. Prior to the advent of ART, various interstitial lung diseases (ILDs) in HIV-infected individuals were characterized as the result of systemic immunological dysregulation, including nonspecific interstitial pneumonitis (NSIP) and lymphocytic interstitial pneumonitis (LIP)2-4. In the current ART period other ILDs are being detected and reported during the consequent stabilization of underlying immunological function2, such as sarcoidosis5, organizing pneumonia (OP), and hypersensitivity pneumonitis (HP).
OP associated with HIV infection is an uncommon entity, as it has been reported only in a few HIV patients so far 6-12. In addition, only one case of usual interstitial pneumonia (UIP) associated with HIV infection has been described in the literature13. In this article, we present an extremely rare case of the co-existence of UIP and OP unmasking undiagnosed HIV infection.
CASE PRESENTATION
A 68-year-old male patient, ex-smoker (60 pack-years), admitted to our Pulmonology Department complaining of dyspnea at exertion and paroxysmal dry cough over the previous three months. His medical history included arterial hypertension, under olmesartan and amlodipine.
Blood pressure was 115/70 mmHg, respiratory rate was 13 breaths/min, heart rate 71 beats per min, body temperature 36.6℃, and oxygen saturation was 93% on room air. Clinical examination revealed only crackles on auscultation at the bases of both lungs, with no other remarkable signs.
Arterial blood gases analysis showed pO2 = 63 mmHg, pCO2 = 33 mmHg, pH = 7.45 and HCO3 = 23 mmol/L, on room air. The chest radiograph revealed reticular opacities in both lungs mostly in the lower lung fields. Laboratory testing including complete blood cell count, biochemistry serum parameters and coagulation testing are presented in Table 1. Finally, electrocardiography did not show any abnormality on admission.
Table 1
Laboratory investigations on admission
[i] ALP: alkaline phosphatase. ALT: alanine aminotransferase. APTT: activated partial thromboplastin time. AST: aspartate aminotransferase. CRP: C-reactive protein. ESR: erythrocyte sedimentation rate. FIB: fibrinogen. GGT: gamma glutamyl-transferase. Hb: hemoglobin. Hct: hematocrit. INR: international normalized ratio. LDH: lactate dehydrogenase. Lym: lymphocytes. Neu: neutrophils. PLTs: platelets. PT: prothrombin time. WBC: white blood cell.
The patient underwent pulmonary function tests (PFTs) which showed forced expiratory volume in 1 second (FEV1) = 2.28 L – 79.39% of predicted, forced vital capacity (FVC) = 2.63 L – 71% of predicted, FEV1/FVC = 86.71, total lung capacity (TLC) = 4.3 L – 66.14% of predicted, residual volume (RV) = 1.42 L – 57.55% of predicted, and diffusion capacity for carbon monoxide (DLCO) = 4.6 mmol/min/kPa – 54.52% of predicted. These results indicated a restrictive lung disease.
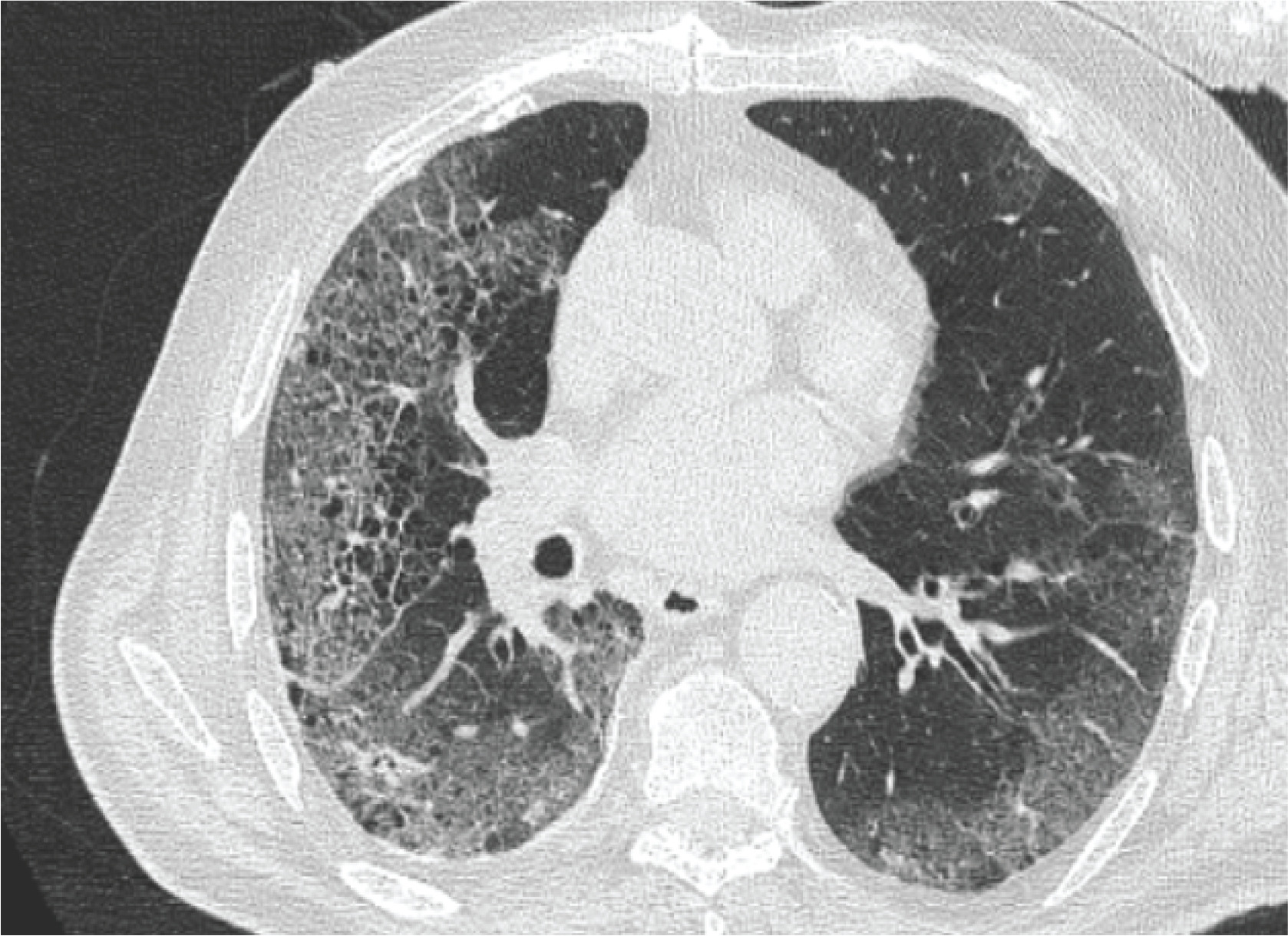
The patient also underwent high resolution computed tomography (CT) of the chest which revealed interlobular septal thickening, ground glass opacities and honeycombing, indicating ILD (Figure 1).
Figure 1
High resolution computed tomography (CT) of the chest revealed interlobular septal thickening, ground glass opacities and honeycombing indicating ILD
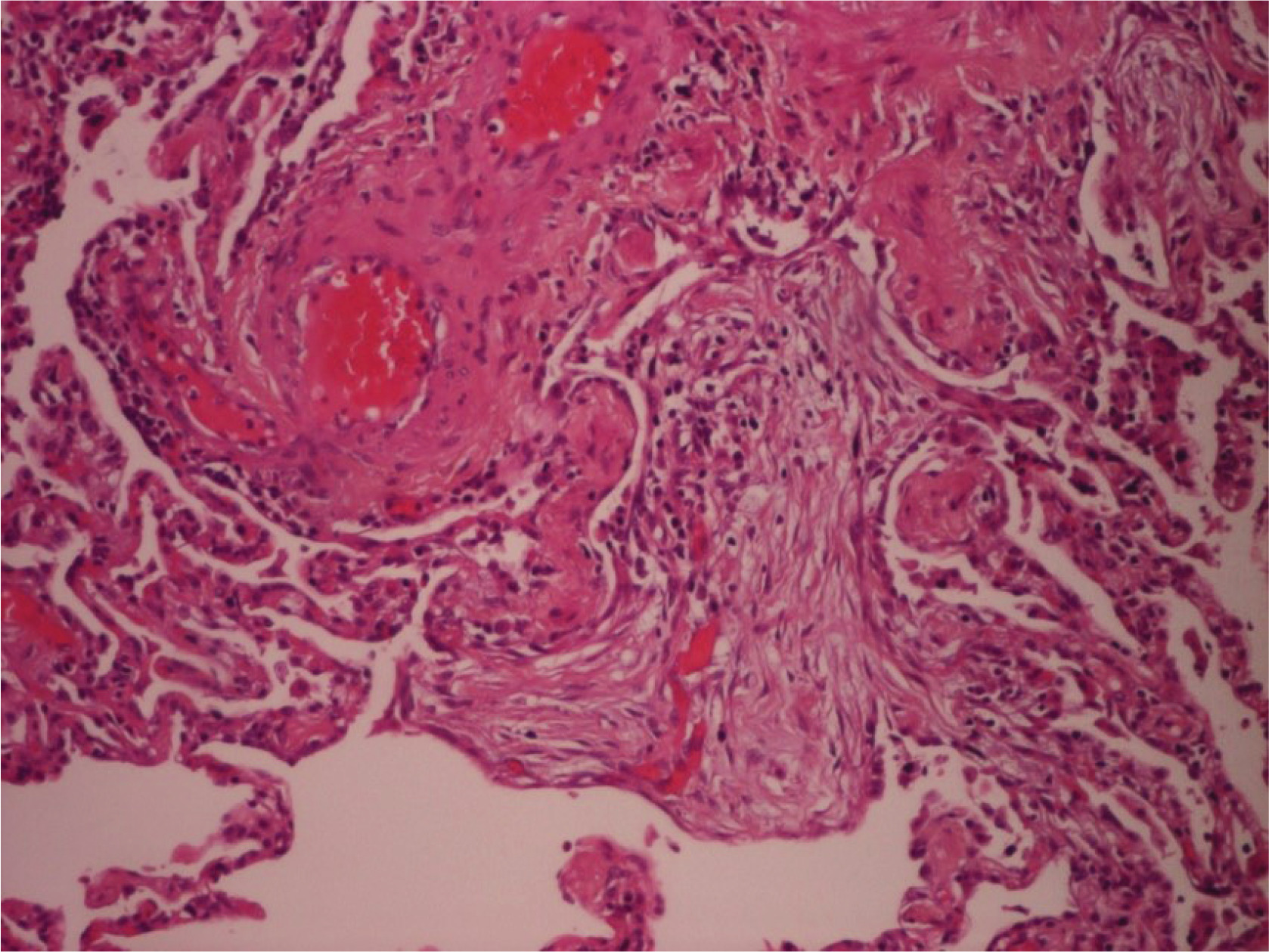
Further laboratory investigation to rule out other underlying autoimmunity diseases was performed (Table 2). The patient was referred to a thoracic surgeon to obtain a lung biopsy for definite diagnosis, as we could not establish an occupational or a connective tissue disorder-related ILD. Lung biopsy was obtained from the left lower lobe with video-assisted thoracoscopic surgery (VATS). The patient was hospitalized in the thoracic surgery clinic for a few days, and he was discharged without complications. Three days later he was admitted again to the hospital with complaints of severe dyspnea and fever. Arterial blood gases analysis showed severe respiratory insufficiency type I (pO2 = 46 mmHg, pCO2 = 29 mmHg, pH = 7.51 and HCO3 = 25 mmol/L) and the patient was hospitalized and received treatment with anticoagulants, broad spectrum-antibiotics, inhaled bronchodilators, intravenous methylprednisolone, and oxygen therapy with Venturi mask delivering 60% oxygen. Due to a progressive respiratory deterioration in the next 15 days, he was intubated and admitted to the intensive care unit (ICU). Surprisingly enough, in the routine evaluation, our patient revealed a HIV test positive, with CD4+ count of 191 cells/mm3. Microbiological examination or respiratory secretions was negative for viral and bacterial infections, including direct fluorescent antibody (DFA) staining for Pneumocystis jirovecii. Unfortunately, he did not respond to corticosteroids and antibiotics, developed ventilator associated pneumonia and passed away due to sepsis and multiorgan failure. Later on, the histopathological examination of the lung biopsy revealed diffuse, interstitial lung disease lesions with UIP peripheral pattern and focal OP lesions in acute phase (Figure 2).
Table 2
Laboratory investigation for underlying autoimmunity
* RF: rheumatoid factor. ANA: antinuclear antibodies. AMA: antimitochondrial antibodies. SMA: smooth muscle antibodies. Anti-ds DNA: anti-double stranded DNA antibodies. cANCA: antineutrophil cytoplasmic antibodies (cytoplasm of ethanol-fixed neutrophils). pANCA: antineutrophil cytoplasmic antibodies (perinuclear cytoplasm). Anti-GBM: antiglomerular basement membrane antibodies. SACE: serum angiotensin converting enzyme.
DISCUSSION
To our knowledge, this is the first report of ILD related to HIV infection with the coexistence of OP and UIP patterns in the pathological examination of a lung biopsy. Our patient exhibited a characteristic ILD presentation with increasing dyspnea, and radiological and physiological features indicative of fibrosis and restrictive lung disease. He had no prior history of respiratory illness and no exposures to the environment or at work that would have been linked to UIP, according to a thorough evaluation of his medical history. His only medications included amlodipine and olmesartan, which have not been associated with ILD.
OP mostly affects the small airways. The exact incidence and prevalence of OP are unknown. Fever, moderate dyspnea, and cough are among the disease’s clinical signs14.
The production of fibrinoid inflammatory cell clusters after epithelial damage, which then leads to intra-alveolar fibrosis, is suggested to represent the etiology of OP. The presence of fibroblastic plugs in the pulmonary bronchioles, alveolar ducts, and alveoli is the pathological hallmark of OP. A biopsy is required for the diagnosis of OP because of these distinctive pathologic findings14. Although VATS lung biopsy is the preferred modality because it is more possible to yield enough tissue, transbronchial lung biopsy is still an option. Broad-spectrum anti-inflammatory drugs, like corticosteroids and immune-modulating macrolides, have a good treatment response because OP is caused by an inflammatory reaction to lung injury. The generation of pro-inflammatory cytokines is inhibited by immune-modulatory macrolides, which are used to treat minor illnesses like OP15,16. There is only one case on the use of macrolide antibiotics in HIV-positive individuals11.
In HIV-positive patients with unexplained chest radiographic lesions, an open or thoracoscopic lung biopsy had a high yield and little morbidity17. When non-invasive methods and bronchoscopic bronchoalveolar lavage (BAL) results are non-diagnostic in patients with substantial pulmonary abnormalities, when the performance of transbronchial biopsy is contraindicated or its result non-diagnostic, and in patients who do not respond to indicated therapy following diagnostic transbronchial biopsy, this modality should be considered12. Furthermore, it is crucial to understand that certain individuals with HIV and assumed P. jirovecii pneumonia who seem to respond to corticosteroid and antimicrobial therapy may experience significant improvement due to the steroid treatment, which can change OP’s course. As a result, OP in the setting of HIV infection might be more common than is currently believed18.
UIP is a pathognomonic mark of idiopathic pulmonary fibrosis (IPF) and has a relentless course with IPF. It is also present in other fibrotic interstitial lung disorders19. Cao et al.13 reported the first case of UIP linked to HIV infection in a 51-year-old HIV patient who was admitted for assessment of identified ILD and complained of fever, dyspnea, exhaustion, anorexia, and gradual weight loss13.
OP may be a precursor to alveolar septal inflammation and may develop into UIP and honeycomb lung. In addition, OP may only be a subsequent inflammatory process to the UIP lesion, which may be the initial lesion, since the existence of both lesions in the same patient, with UIP as the primary and OP as the secondary lesion, has previously been described in non-HIV patients with rheumatic diseases20.
We should mention that several case reports have connected acute exacerbation of ILD (AE-ILD) to both bronchoscopic and surgical procedures. While initial descriptions of AE following biopsy primarily focused on IPF patients, it is now evident that AE can manifest in various ILDs subsequent to biopsy procedures21,22.
Our patient probably exhibited AE-ILD and a very fulminant course of the disease that did not allow reasonable follow-up. The post-exacerbation mortality in ILDs is reported to range from 33–83%23,24.
The co-existence of OP with UIP represents a distinctive and intricate pattern in pulmonary pathology. This juxtaposition of two distinct entities within the lung parenchyma poses challenges in both diagnosis and management. The convergence of these patterns signifies a complex interplay between inflammatory and fibrotic processes within the lung. Histopathological examination becomes crucial for delineating the features of OP and UIP in this co-existence scenario. The identification of organizing pneumonia, with its characteristic buds of granulation tissue within the alveolar spaces, alongside the fibrotic changes typical of UIP, requires meticulous scrutiny. Additionally, radiological imaging, such as HRCT, can aid in visualizing the distinct patterns and their distribution within the lungs25,26.
CONCLUSION
The coexistence of OP and UIP in HIV patients is a very rare entity; however, clinicians should be aware of this condition. Although noninfectious pulmonary disorders are less frequent than infectious in HIV patients, the diagnosis of different types of ILDs should be considered after exclusion of infections and malignancies.