


INTRODUCTION
The crazy paving pattern ‘consists of scattered or diffuse ground-glass attenuation with superimposed interlobular septal thickening and intralobular lines’, resembling a street pavement1. It was first described in pulmonary alveolar proteinosis (PAP), but it is not currently considered as a pathognomonic sign of this condition, as it can be found in a variety of disorders, including malignancy, infection, hemorrhage, cardiogenic pulmonary oedema, acute eosinophilic pneumonia, hypersensitivity pneumonitis (HP), sarcoidosis, and rarely nonspecific interstitial pneumonia (NSIP). Thus, the diagnosis of a patient with this pattern on chest imaging needs to be based on clinical and/or histological data2. However, even after a comprehensive evaluation, a percentage of patients with interstitial lung disease (ILD) on chest high-resolution computed tomography (HRCT) cannot be given a specific diagnosis, and may be labelled as unclassifiable ILD (uILD)3,4. Based on cohorts recruited from ILD referral centers, the prevalence of uILD is approximately 12%, but with substantial variability among studies, due to the heterogeneity of study design and diagnostic approaches across several ILD cohorts5.
CASE PRESENTATION
A woman aged 76 years, never smoker, with a medical history of hypothyroidism, gastroesophageal reflux disease, well-regulated arterial hypertension, and dyslipidemia, was referred due to shortness of breath and mild fatigue over the past six months. She claimed exposure to mold, while chronic medication included ramipril, atorvastatin, levothyroxine, and esomeprazole.
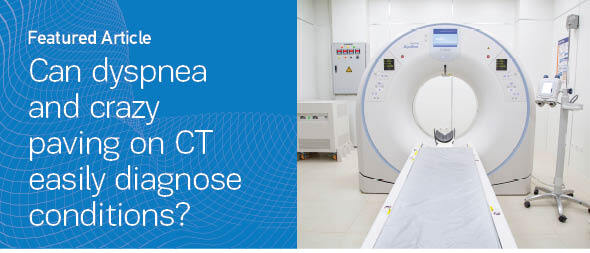
Two consecutive HRCTs were performed with an interval of six months, and both showed diffuse alveolar-type infiltrates and areas of ground glass opacities (GGOs), with thickened interlobular septa and intralobular lines, resembling the so-called crazy paving pattern (Figures 1A–1D). At that moment, the patient was referred to our department for further investigation.
Figure 1
Lung window of serial chest HRCT scans (A–B: first scan; C–D: second scan after 6 months; E–F: third scan 2 months later) of the patient showing geographic areas of ground-glass opacification associated with smoothly thickened interlobular septa in both lower lobes. This appearance is called the ‘crazy paving’ pattern. Radiological findings were deteriorating over the time, with the appearance of bronchial distortion more prominent in the last scan (E–F)
On admission, the clinical examination revealed inspiratory fine crackles in both lung bases, rhythmic heart sounds, soft and non-tender abdomen with present bowel sounds, and absence of peripheral edema or jugular venous distention. She had normal body temperature (36.5oC), normal arterial pressure (130/70 mmHg), and a respiratory rate of 18 breaths per minute. Arterial blood gas (ABG) measurement on room air revealed hypoxemia, particularly pO2: 59 mmHg, pCO2: 32 mmHg, pH: 7.46, and HCO3: 22 mEq/L. Pulmonary function tests (PFTs) revealed a restrictive pattern with forced vital capacity (FVC) of 65% of predicted value (1610 mL), forced expiratory volume in one second (FEV1) to FVC ratio of 85%, and severely reduced diffusing capacity for carbon monoxide (DLCO), 30% of predicted value.
Electrocardiography (ECG) revealed no apparent signs of ischemia. Transthoracic heart ultrasound was indicative of heart failure with preserved ejection fraction (diastolic dysfunction). Although the patient was lacking clear clinical or radiological evidence of heart failure, she received mild diuresis without any improvement.
She also underwent complete laboratory testing, including immunological testing (values within normal range for complement C3 and C4 and immunoglobulins IgA, IgG, and IgM, as well as negative results for rheumatoid factor, antinuclear antibodies, anti-cyclic citrullinated protein antibodies, anti-double stranded DNA antibodies, anti-neutrophil cytoplasmic antibodies, and anti-extractable nuclear antigen antibodies), while rheumatologic evaluation did not detect any signs of systemic autoimmune rheumatic disease (SARD). Inflammatory markers were not found to be increased (White blood cells: 7.5 K/μL, C-reactive protein: 0.99 mg/dL, and erythrocyte sedimentation rate: 31 mm/h), while there was neither hepatic nor renal dysfunction or hematuria. Moreover, eosinophils consisted of 975 cells/μL, immunoglobulin IgE was at 531 IU/mL and radioallergosorbent testing was positive for house dust mites.
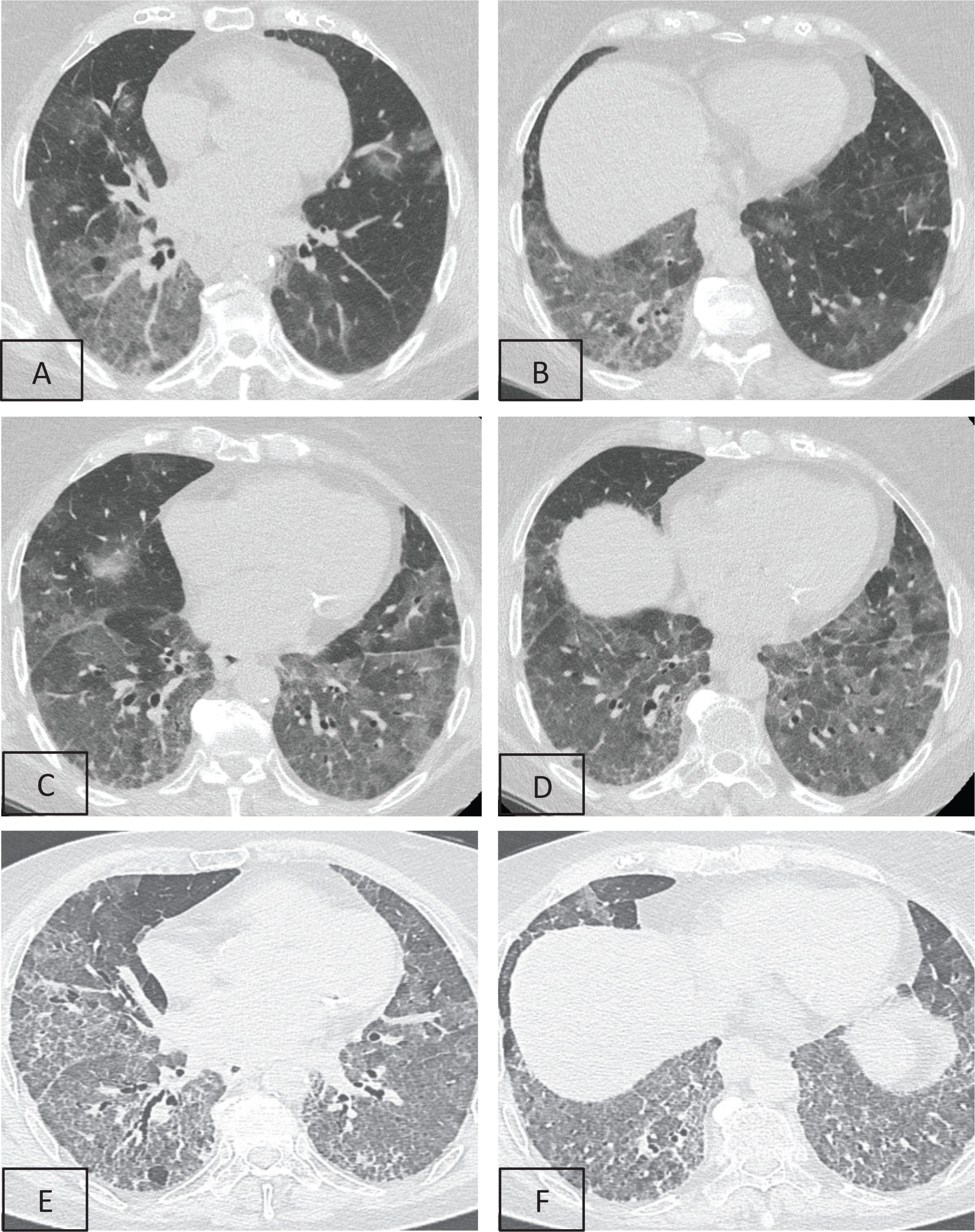
The differential diagnosis so far included SARD with pulmonary involvement, HP due to mold exposure, and PAP due to consistent chest imaging. Therefore, bronchoscopy was performed, which revealed no intraluminal lesions. Washing cultures for common pathogens, immunofluorescence for Pneumocystis jirovecii, and Ziehl-Neelsen staining were all negative. The cell populations in the bronchoalveolar lavage (BAL) were 70% macrophages, 18.5% neutrophils, 8.5% eosinophils, and 3.5% lymphocytes. Bronchial cytology was negative for malignancy. Nevertheless, periodic-acid-Schiff (PAS) stain of BAL revealed positive deposits of acellular granulocytic eosinophilic material, and this led to the provisional diagnosis of PAP (Figure 2).
Figure 2
PAS stain of BAL cells that revealed positive deposits of acellular granulocytic eosinophilic material
Based on this BAL finding, the patient was transferred to another Hospital for administration of inhaled recombinant granulocyte-macrophage colony-stimulating factor (GM-CSF)6,7, given an overt deterioration of oxygen saturation (the patient needed high flow nasal oxygen therapy), while waiting for the antibodies against GM-CSF and the genetic test for congenital PAP. A second bronchoscopy was also performed. The result of the autoantibody testing, genetic testing, and the PAS stain of the second BAL were all negative, making the diagnosis of PAP finally unlikely.
Therefore, the patient received broad spectrum antibiotics, as well as treatment for P. jirovecii. New PFTs after antibiotic treatment revealed further worsening (FVC: 55% and DLCO: 20% of predicted values), while a third chest HRCT was also remarkable for further deterioration (Figures 1E and 1F). New immunological testing, including myositis specific antibodies, was found between normal limits. The second provisional diagnosis was HP. However, the patient was deteriorating during this time frame of 4 months, despite the abolition of the exposure to mold, while there was neither the characteristic three density sign on HRCT nor BAL lymphocytosis8. Thus, by excluding other possible diagnoses, and following a multidisciplinary discussion (MDD), the patient was labelled as uILD, and started treatment with per os corticosteroids (gradually tapered), and mycophenolate mofetil (MMF) as a corticosteroid-sparing agent. Furthermore, based on the current guidelines about progressive pulmonary fibrosis9, nintedanib was added. The patient three months later is improved (Table 1), without need for supplemental oxygen, and she is carefully monitored. She has not presented any systematic signs and symptoms yet; thus, it remains unknown whether the condition will evolve to SARD.
Table 1
Pulmonary function tests of the patient, at different time points
DISCUSSION
Unclassifiable ILD is variably defined in the literature, and the standardization of the nomenclature and definition for this group of patients remains elusive3,10,11. The most common definition of uILD is the absence of a specific diagnosis following an MDD and review of available clinical, radiological, and pathological information10,12,13. According to current literature, uILD is associated with a HRCT pattern indeterminate for usual interstitial pneumonia (UIP), or most consistent with non-IPF diagnosis4,12,14. In our case, HRCT findings were indeterminate for UIP. GGOs on HRCT had a geographic distribution and were combined with smoothly thickened interlobular septa within areas of air space disease, whilst no bronchial dilatation was present.
This finding could be attributed to PAP, a disorder characterized by surfactant, lipid and protein accumulation in the alveolar spaces, which has either autoimmune, secondary, or congenital etiology15,16. Autoimmune PAP compromises 90% of all cases and is a result of anti-GM-CSF antibodies that lead to GM-CSF deficiency. GM-CSF is an essential cytokine for alveolar macrophages development and function. Secondary PAP is characterized by dysfunctional alveolar macrophages secondary to hematological, infectious, or environmental factors16. However, there was no evidence supporting secondary PAP in our patient. PAS staining is used for detection of structures that contain high concentrations of glycogen, glycoprotein, and proteoglycans, therefore the presence of PAS positive flocculent proteinaceous material in BAL can support PAP diagnosis17. However, PAS positive staining has been described also in BAL cells in other interstitial lung diseases (ILDs), including idiopathic pulmonary fibrosis (IPF), HP and sarcoidosis18, whilst PAS positivity has been detected in fungal infections as well19. On the other hand, the presence of IgG anti-GM-CSF antibodies is considered highly sensitive and specific for autoimmune PAP, as supported also by a recent study which detected 100% sensitivity and 98% specificity using a cut-off level of 2.8 μg/mL20. Therefore, negativity of anti-GM-CSF antibodies, finally excluded PAP as a diagnosis in our case.
Furthermore, HP criteria were not met8, and SARD was not supported neither by serology markers nor by clinical assessment and rheumatologic evaluation. Histological evaluation could not be achieved, due to the severe pulmonary function impairment, as well as the refusal by the patient. An MDD that includes pulmonologists, radiologists and pathologists undoubtedly assists in reaching a consensus in ILD diagnosis, since the proportion of uILD patients seem to be lower in cohorts that have undergone an MDD5. Thus, in our case, following MDD, a diagnosis of uILD was reached.
The management of patients with uILD remains challenging, given the plethora of uncertainties21,22. Immunosuppressive medication may be an option when there is a low likelihood of IPF and a primary differential diagnosis of HP, SARD-ILD, or other non-IPF ILD4,11,21,22. GGO and the absence of honeycombing on HRCT can drive the clinician’s decision for immunomodulatory treatment. BAL lymphocytosis has been also associated with better outcomes in patients treated with anti-inflammatory drugs23. However, given the absence of BAL lymphocytosis, as well as the progressive phenotype9 , and the baseline lung function, which has been associated with worse outcomes4, the addition of the antifibrotic agent nintedanib was decided in our case. Based on recently published cohorts, patients with uILD might present with a progressive fibrotic phenotype in more than 20% of cases24-26. Pirfenidone has been also evaluated in patients with uILD, slowing the rate of FVC decline, although the primary end-point of home-based spirometry was not met in this study26. More consistently defined patients’ groups and more careful selection of trial endpoints may be required to overcome the difficulty concerning the heterogeneous population and biology of uILD.
CONCLUSION
The differential diagnosis of crazy paving pattern on HRCT is broad and challenging. In cases where a definite diagnosis cannot be reached, even following an MDD, the patient may be provided with the uILD diagnosis. Treatment options of uILD are challenging and require a case-by-case consideration of the relative likelihoods of the differential diagnosis, the anticipated disease behavior, and medication adverse effects and tolerability. Finally, it is worth highlighting that patients with uILD should be closely monitored for future development of SARD. Future studies and research tools are needed to better define ILD subgroups. Advances in molecular phenotyping with transcriptomics, proteomics, metabolomics, and epigenetics may eventually allow an accurate ILD diagnosis in some patients who are currently considered unclassifiable11,27.