



INTRODUCTION
Interstitial lung diseases (ILD) constitute a heterogeneous group of diseases that may occur in the contexts of systemic disorders, mainly collagen vascular diseases (CVD), and lung toxicity secondary to inhaled agents or drugs, and may also be of unknown origin (idiopathic). Classification of ILD requires histologic, clinical, and radiological patterns setting the diagnosis by a multidisciplinary approach. The classification includes major idiopathic interstitial pneumonias (idiopathic pulmonary fibrosis, idiopathic nonspecific interstitial pneumonia, respiratory bronchiolitis – interstitial lung disease, desquamative interstitial pneumonia, cryptogenic organizing pneumonia, acute interstitial pneumonia), rare idiopathic interstitial pneumonias (idiopathic lymphoid interstitial pneumonia, idiopathic pleuroparenchymal fibroelastosis) and unclassifiable ones1. When they occur as a pulmonary manifestation of CVD, they may overlap with other CVD-related respiratory disorders. We present the case of a young male patient with NSIP occurring in the context of primary Sjogren syndrome, recurrent pulmonary embolism, and pulmonary hypertension.
CASE PRESENTATION
A 28-year-old black male, a refugee from Sub-Saharan Africa and a non-smoker, was admitted because of shortness of breath, dry cough, and thoracic pain for the last two weeks. His medical history was significant for asthma since childhood, hepatitis B, pulmonary tuberculosis for which he received full treatment six years ago, pulmonary embolism, and poorly defined interstitial pneumonia diagnosed two years ago. His medication included inhaled salmeterol/budesonide and occasional use of methylprednisolone, Rituximab, and acenocumarol with poor compliance. In 2020, he had his first episode of pulmonary embolism. At that time, computed tomography (CT) of the lungs also revealed interstitial pneumonia. The patient reported no inhalation of toxic agents or any drug use that could provoke lung fibrosis. The laboratory tests were positive for ΑΝΑ, anti-Ro52, and anti-ENA. Then, the diagnosis of systemic lupus erythematosus (SLE) was made and the patient was treated with a high dose of corticosteroids and Rituximab. It should be noted that after hospital discharge, the patient did not receive the suggested treatment regularly. Over the past two years, the patient reports multiple hospitalizations for shortness of breath with unclear diagnosis and treatment.
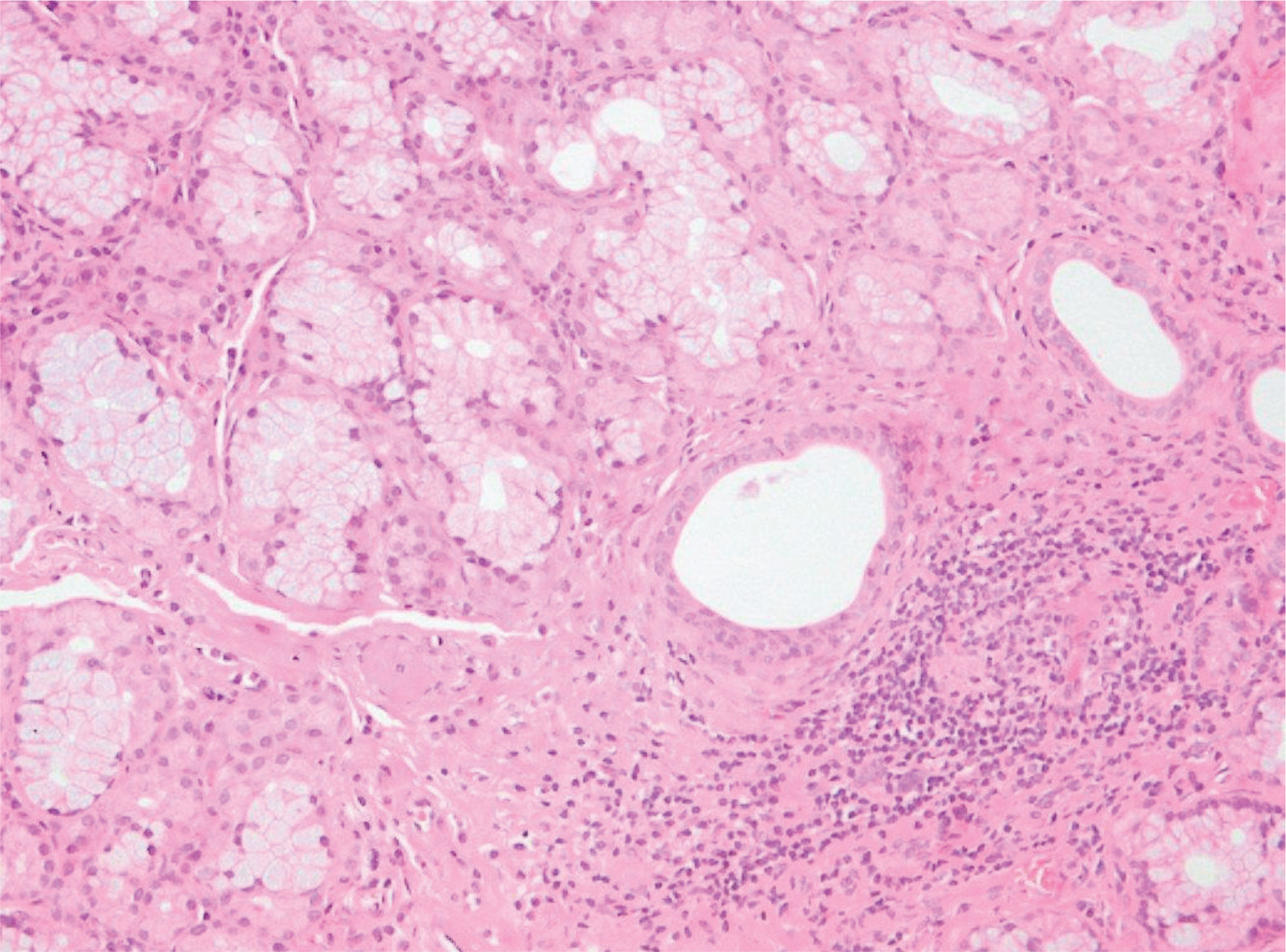
Upon admission to our department, his temperature was 37°C, his respiratory rate was 32/min, his blood pressure was 110/80 mmHg, his heart rate was 105/min, and the SpO2=89% while breathing room air. Lung auscultation did not reveal crackles. He did not have finger clubbing. Arterial Blood Gas (ABG) testing demonstrated pO2=57 mmHg, pCO2=31 mmHg, pH=7.44, and HCO3=23 mEq/L. Chest x-ray showed reticulation in both lungs. The computed tomography pulmonary angiography (CTPA) scan demonstrated a new episode of pulmonary embolism and bilateral patchy ground-glass attenuation, reticulation, and traction bronchiectasis, with middle and lower lung zone distribution (Figure 1). Treatment with anticoagulant was restarted. The C-reactive protein (CRP) was 4 mg/dL (normal range: <0.5 mg/dL). The heart ultrasonography showed a hypertrophic right ventricle, D-shaped middle-ventricular wall, and pulmonary hypertension [right ventricular systolic pressure (RSVP) = 60 mmHg]. Bronchoscopy performed after two weeks, to exclude lung infection, showed no intrabronchial lesions and bronchoalveolar lavage cultures came out negative for bacteria, M. tuberculosis, Aspergillus, and Nocardia. Also, the HIV test came out negative. The serologic studies demonstrated ANA (+), ENA (+), and anti-RO52 (+), while antisynthetase syndrome antibodies came out negative. Given the patient’s laboratory and clinical features were not compatible with the diagnosis of SLE and since the patient reported the presence of xerophthalmia and xerostomia, primary Sjogren syndrome was suspected and a biopsy of salivary glands was performed. Salivary gland histology depicted lymphocytic cell infiltration (50 lymphocytic foci/4 mm2), establishing the diagnosis of Sjogren syndrome (Figure 2).
Figure 1
Computed tomography of the lungs showing patchy ground-glass attenuation, reticulation, and traction bronchiectasis
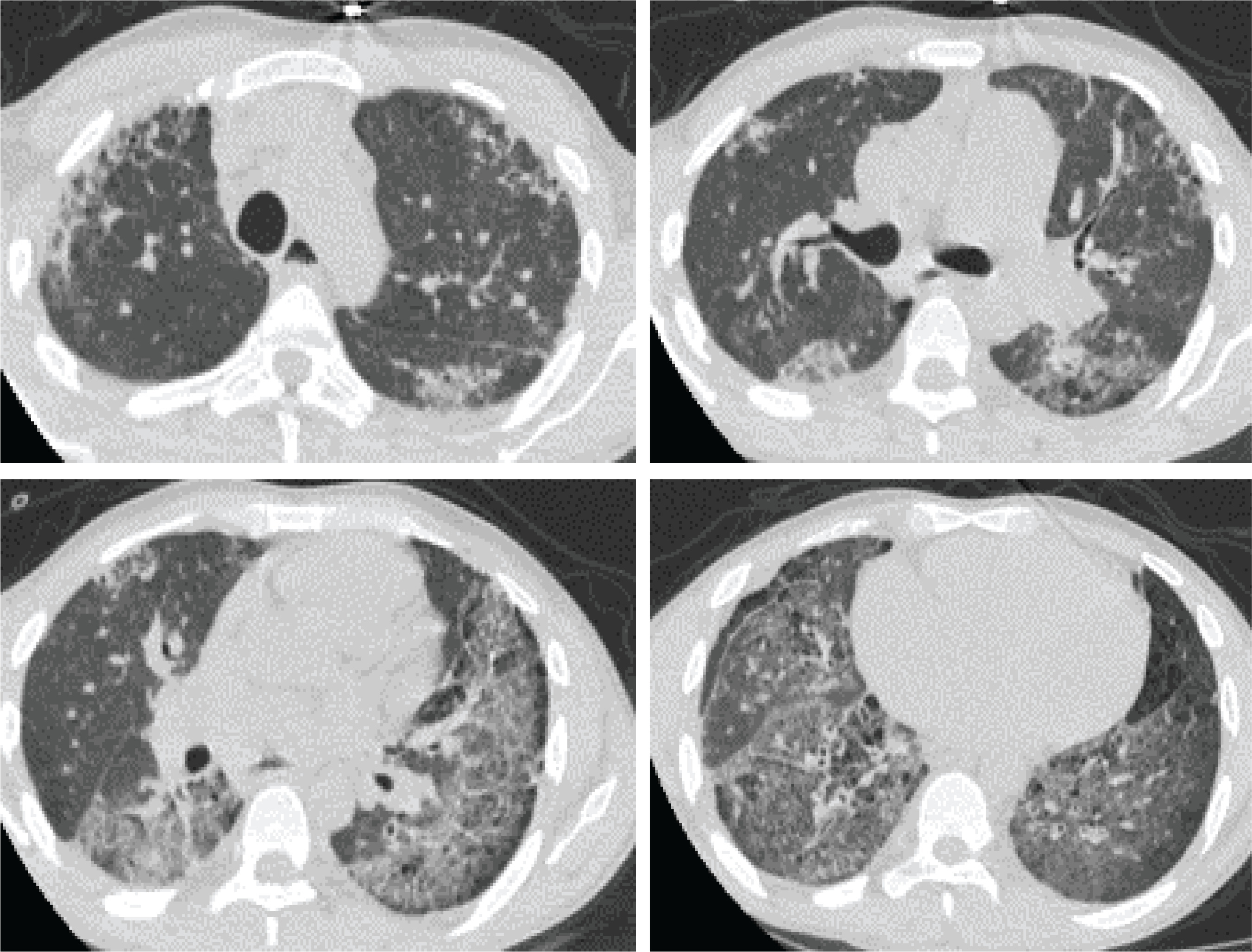
Figure 2
Photomicrograph of salivary gland biopsy showing lymphocytic infiltration (eosin-hematoxylin stain, ×200)
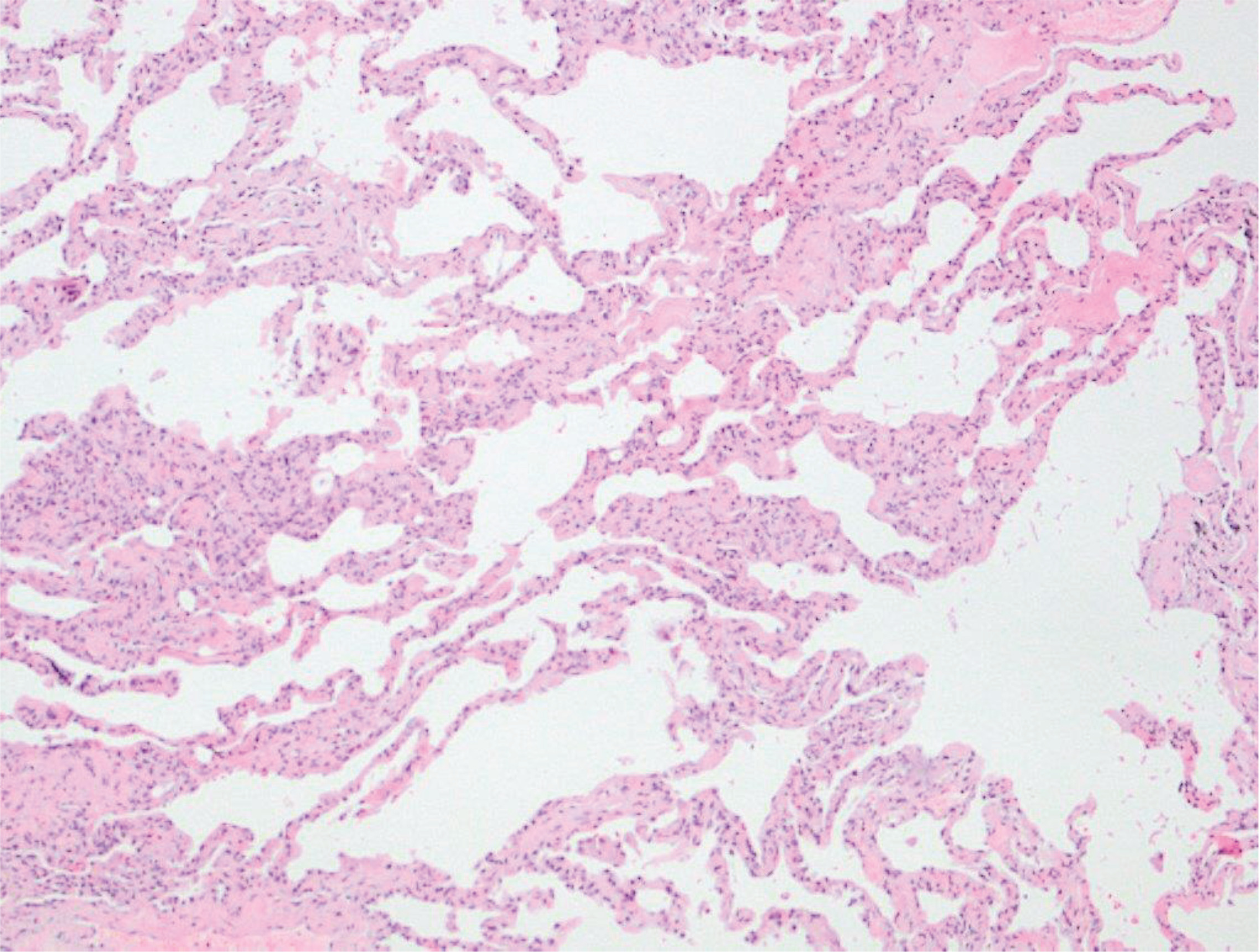
Pulmonary function tests revealed significantly decreased lung diffusion capacity (DLCO: 2% of predicted) and restrictive lung disorder [FEV1/FVC: 71%; FVC: 1990 mL (38% of predicted), FEV1: 1540 mL (45% of predicted), TLC: 4330 mL (62% of predicted)]. A thoracoscopic lung biopsy was performed and revealed cellular non-specific interstitial pneumonia (Figure 3). He was then given prednisone 1 mg/kg body weight per day. Mycophenolate mofetil was added to permit prednisone dose tapering.
Figure 3
Photomicrograph of lung biopsy showing mild alveolar septal thickening without fibrosis and infiltration of inflammatory cells. Lung architecture is preserved (eosin-hematoxylin stain, ×100)
One month after treatment initiation, pulmonary function tests were significantly improved (FVC: 62% of predicted, FEV1: 62% of predicted, DLCO: 15% of the predicted) but the respiratory failure persisted (ABG: PO2=54; PCO2=34; pH=7.42; HCO3=23). Moreover, the patient underwent a 6-minute walk test (6MWT) passing through 300 meters without feeling tired or short of breath. CTPA showed the resolution of pulmonary clots. A repeat heart ultrasonography, two months after the commencement of anticoagulants, showed no improvement in pulmonary hypertension. A transesophageal echocardiography, to exclude valvular abnormalities, was performed. Ventilation–perfusion scan was inconclusive for chronic thromboembolic pulmonary hypertension (CTEPH), probably due to lung scarring and bilateral ground glass opacities. Thus, having passed more than 3 months after the initiation of anticoagulants and given a persisting abnormal cardiac triplex, right-heart catheterization was conducted demonstrating pulmonary atrial pressure, 2 mmHg; pulmonary artery pressure, 46/24/31 mmHg (systolic/diastolic/mean); pressure wedge, 6 mm Hg; cardiac output, 4.5 L/min; cardiac index, 2.4 L/min/m2; and pulmonary vascular resistance, 5.5 Wood units. Since the patient was already on rivaroxaban, vasodilative treatment (macitentan and tadalafil) was initiated. After 3 months of full treatment, there was an improvement in pulmonary hypertension (RSVP = 44 mmHg) and 6MWT (460 m without shortness of breath).
DISCUSSION
Primary Sjogren syndrome (pSS) is a multisystem autoimmune rheumatic disease characterized by female predominance and a prevalence in Europe of 0.1–4.8%2,3. Clinical manifestations range from localized glandular disease to complex and even life-threatening systemic features affecting musculoskeletal, skin, neurological, cardiovascular, and pulmonary systems. The laboratory findings in patients with extra glandular manifestations depend on the degree of systemic inflammation and specific organ involvement. Serum autoantibodies are present such as antinuclear antibodies (ANA>1:320), anti-Ro/SSA, anti-La/SSB, centromere antibodies, anti-citrullinated peptide antibodies, and rheumatoid factor2. The ACR/EULAR classification criteria include labial salivary gland with focal lymphocytic sialadenitis and focus score ≥1/mm2, positive anti-SSA (Ro) antibodies (each scoring 3), Schirmer test ≤5 mm/5 min on at least one eye and abnormal ocular staining score >5 (each scoring 1). Individuals with a total score ≥4 of the items above meet the Sjogren syndrome criteria4. Sjogren syndrome may affect the upper respiratory tract as well as large and small airways and also causes cystic or interstitial lung disease (prevalence of about 20%)5. ILD has several forms including non-specific interstitial pneumonitis (NSIP), usual interstitial pneumonitis (UIP), lymphocytic interstitial pneumonitis (LIP), and cryptogenic organizing pneumonia (COP). In our case, the form of ILD is a cellular pattern of NSIP, which carries a better prognosis than the fibrosing pattern. The optimal treatment for this kind of case is glucocorticoids in high doses, immunosuppressive drugs, and nintedanib in progressive fibrotic ILD5. The incidence of venous thromboembolism (VTE) in pSS is notably increased; this might be explained by impairment of the fibrinolytic system and reduced activity of intrinsic anticoagulant agents due to inflammation observed in pSS6. Pulmonary hypertension is an extremely severe and rare complication of Sjogren syndrome. It is mostly related to vasculitis (prolonged vasospasm, structural vessel remodeling, and irreversible thrombotic obstruction of pulmonary arterioles), B-cell activation, and autoimmunity7. It is also more frequently associated with the Raynaud phenomenon, antinuclear, anti-Ro/SSA, anti-RNP autoantibodies, positive rheumatoid factor, and hypogammaglobinemia8. The mortality risk is significantly high, with lymphoma and cardiovascular diseases being the leading causes of death9. In our case, it was difficult to decide if we face a case of chronic thromboembolic pulmonary hypertension or whether pulmonary hypertension occurred as a manifestation of Sjogren syndrome. However, since the patient was going to receive life-long anticoagulant treatment because of two unprovoked pulmonary embolisms, we decided to add pulmonary vasodilators.
CONCLUSION
This is an uncommon case of non-specific interstitial pneumonitis, pulmonary embolism, and pulmonary hypertension in a patient with Sjogren disease. Proper treatment of ILD led to significant pulmonary function test improvement. However, the persistence of respiratory failure suggested additional pathology which proved to be pulmonary hypertension. We conclude that in a patient with systemic disorders, the possibility of pleomorphic respiratory involvement should be considered.