

INTRODUCTION
Systemic sclerosis or scleroderma (SSc) is a systemic inflammatory disorder with features including microangiopathy, inflammation, and fibrosis of the skin and internal organs1. Pulmonary involvement occurs in more than 80% of patients with scleroderma, most frequently interstitial lung disease (ILD)2. ILD is a group of diseases consisting of chronic inflammation within the lung and varying degrees of lung fibrosis. ILD is a variable manifestation of scleroderma and is known as ILD associated with scleroderma (SSc-ILD)3. Generally, ILD develops simultaneously or following diagnosis of a connective tissue disease (CTD), but ILD can also precede CTD establishment4. ILD appearing as the first manifestation is most commonly observed in idiopathic inflammatory myopathies, sometimes in rheumatoid arthritis patients, but rarely in scleroderma patients5. This case will explore a patient with ILD preceding the diagnosis of scleroderma.
CASE PRESENTATION
A 52-year-old female non-smoker presented with shortness of breath on exertion and chronic cough for 1 year. Patient had a history of ILD dating back 5 years and recent abnormal chest x-ray raising a suspicion of pulmonary fibrosis. Patient was given salbutamol 100 μg to take as needed, but did not find that it helped her symptoms. Previous medical history includes breast cancer (ductal carcinoma in situ) and right thoracotomy for neurilemmoma. Patient was positive for estrogen receptor (ER) and progesterone receptor (PR), but negative for human epidermal growth factor receptor 2 (HER2). Patient underwent right breast lumpectomy followed by mastectomy and did not undergo chemotherapy or radiation.
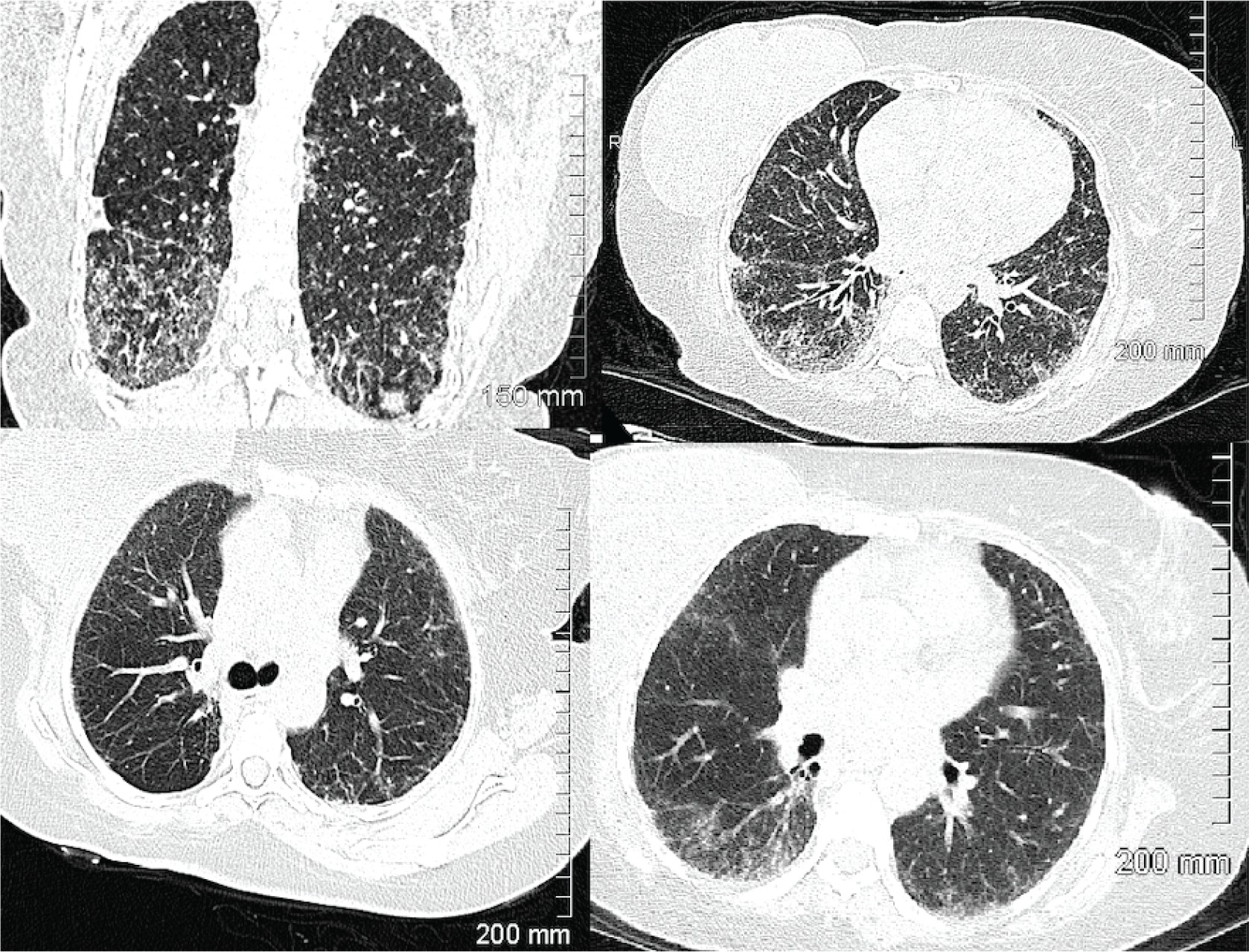
Physical examination revealed bibasilar crackles with no wheezing or prolonged expiratory phase. There was no evidence of cyanosis, clubbing or edema. Vital signs were normal with an oxygen saturation level of 99% at rest. A high-resolution computed tomography (HRCT) chest scan was ordered and was very abnormal showing significant evidence of interstitial lung disease compared to a CT done 5 years prior (Figure 1).
Figure 1
HRCT scan of chest at time of initial consultation. Findings compared to images done 5 years prior and suggested progression of ILD, likely fibrotic NSIP. Septal thickening and ground glass opacities present in both lungs, with associated bronchiectasis in lower lungs
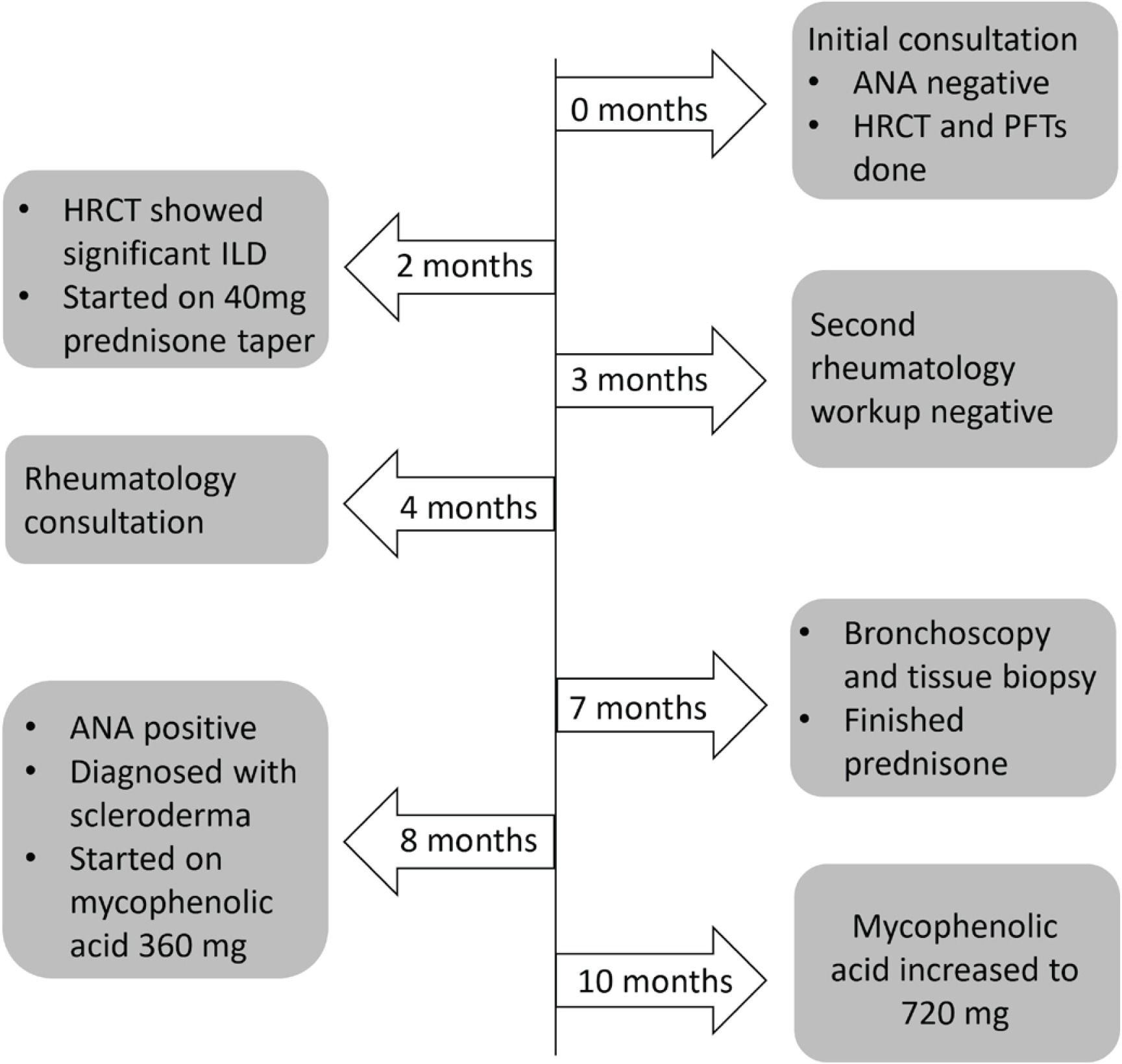
Pulmonary function tests (PFTs) were done and showed severe restrictive lung disease with forced vital capacity (FVC) of 1.25 L (45% predicted), forced expiratory volume in 1 second (FEV1) of 1.17 L (51% predicted), and very severely reduced diffusion capacity of carbon monoxide (DLCO) of 12% (Table 1). A rheumatology workup to rule out underlying rheumatologic etiology was done which was negative (Table 2). The patient was started on prednisone 40 mg with a tapering dose for approximately 5 months (Figure 2). A bronchoscopy and tissue biopsy were ordered but held off when patient showed improvement in symptoms. Bronchoscopy and tissue biopsy chosen versus open lung biopsy due to severely reduced DLCO.
Table 1
Results of pulmonary function tests (PFTs) in relation to time after initial visit
| Time | FVC (L) | FVC % predicted | FEV1(L) | FEV1 % predicted | DLCO |
|---|---|---|---|---|---|
| 2 months | 1.25 | 45 | 1.17 | 51 | 12 % |
| 4 months | 1.34 | 49 | 1.20 | 52 | N/D |
| 6 months | 1.23 | 51 | 1.09 | 54 | 25 % |
| 7 months | 1.20 | 44 | 1.07 | 47 | N/D |
| 8 monthsa | 1.18 | 43 | 0.96 | 41 | N/D |
| 10 months | 1.26 | 46 | 1.09 | 46 | N/D |
| 12 months | 1.13 | 41 | 0.97 | 43 | N/D |
| 1 year 7 months | 1.11 | 43 | 1.00 | 47 | 10 % |
| 2 years 5 months | 0.76 | 32 | 0.76 | 39 | N/D |
| 2 years 10 months | 0.63 | 23 | 0.55 | 25 | N/D |
| 3 years 3 months | 0.64 | 25 | 0.61 | 29 | N/D |
| 3 years 9 months | 0.60 | 24 | 0.45 | 21 | N/D |
| 5 years 3 monthsb | 1.87 | 80 | 1.45 | 79 | 41 % |
| 6 years 0 monthsc | 1.76 | 77 | 1.37 | 76 | 45 % |
Table 2
Rheumatology laboratory results in relation to time after initial visit
| Tests | 0 months | 3 months | 8 months |
|---|---|---|---|
| ESR | 14 mm/h | N/D | N/D |
| HSCRP | 4.8 mg/L | N/D | N/D |
| RF | Negative | N/D | Negative |
| ANA | Negative | N/D | Positive > 1:640 |
| Anti-ENAa | N/D | Negative | Negative |
| Complement C3 | N/D | 1.15 g/L | 1.18 g/L |
| Complement C4 | N/D | 0.24 g/L | 0.26 g/L |
| Anti-ds DNA | N/D | Negative | Negative |
The patient had a rheumatology consultation concerning discoloration in hands and symptoms of carpel tunnel syndrome. Patient described noticing discoloration in hands for approximately 2 years, as well as numbness and tingling in hands that at times would radiate into elbows. Complained of pain in bilateral CMC joints, along with the 2nd and 3rd PIPs bilaterally. Clinical examination revealed Raynaud’s phenomenon without other associated signs and symptoms of scleroderma.
A second rheumatology workup was ordered and was unrevealing. A bronchoscopy and tissue biopsy were scheduled when dyspnea and cough returned following tapering of prednisone. Biopsy showed no evidence of malignancy on LN7 and 10R, and pathology results from bronchoscopy showed benign bronchiole and alveolar tissue. Bronchial washings of the right lower lung were negative for malignancy. The patient was referred for a thoracic surgery consultation regarding open lung biopsy to help assist in making a diagnosis. A six-minute-walk test was also completed, showing significant desaturation upon exercise (Table 3). Blood gas analysis had results of PaO2 44mmHg and PaCO2 82mmHg.
Table 3
Six-minute-walk test results in relation to time after initial visit
| Tests | 8 months | 1 year 7 months | 4 yearsa |
|---|---|---|---|
| Time walked | 6 minutes 0 seconds | 6 minutes 20 seconds | 6 minutes 0 seconds |
| Distance walked | 331 m | 404 m | 305 m |
| SpO2 at rest | 96% | 96% | 98 % |
| SpO2 minimum | 84% | 84% | 86 % |
| Duration with SpO2 below 89% | 2 minutes 50 seconds | 4 minutes 0 seconds | 1 minute 0 seconds |
| BORG dyspnea upon completion | 1 | 2 | 5 |
As illustrated in the timeline in Figure 2, autoimmune serologies were repeated when new rheumatologic symptoms arose, and respiratory symptoms worsened. Patient complained of pain in basal thumb region and had a tender lesion in the index finger pulp along with dilated nailfolds, that were not initially present. Clinical examination now also revealed slight puffiness in the fingers and ankles and CMC joint tenderness. Results came back positive for antinuclear antibody (ANA) with nucleolar pattern suggestive of scleroderma, compared to a negative result 8 months prior. Diagnosis of scleroderma was made based on ANA >1:640, Raynaud’s phenomenon, puffy fingers, dilated nailfolds, finger lesion, and history of ILD. The patient was started on mycophenolic acid 360 mg twice daily (later increased to 720 mg) for her new diagnosis of SSc-ILD.
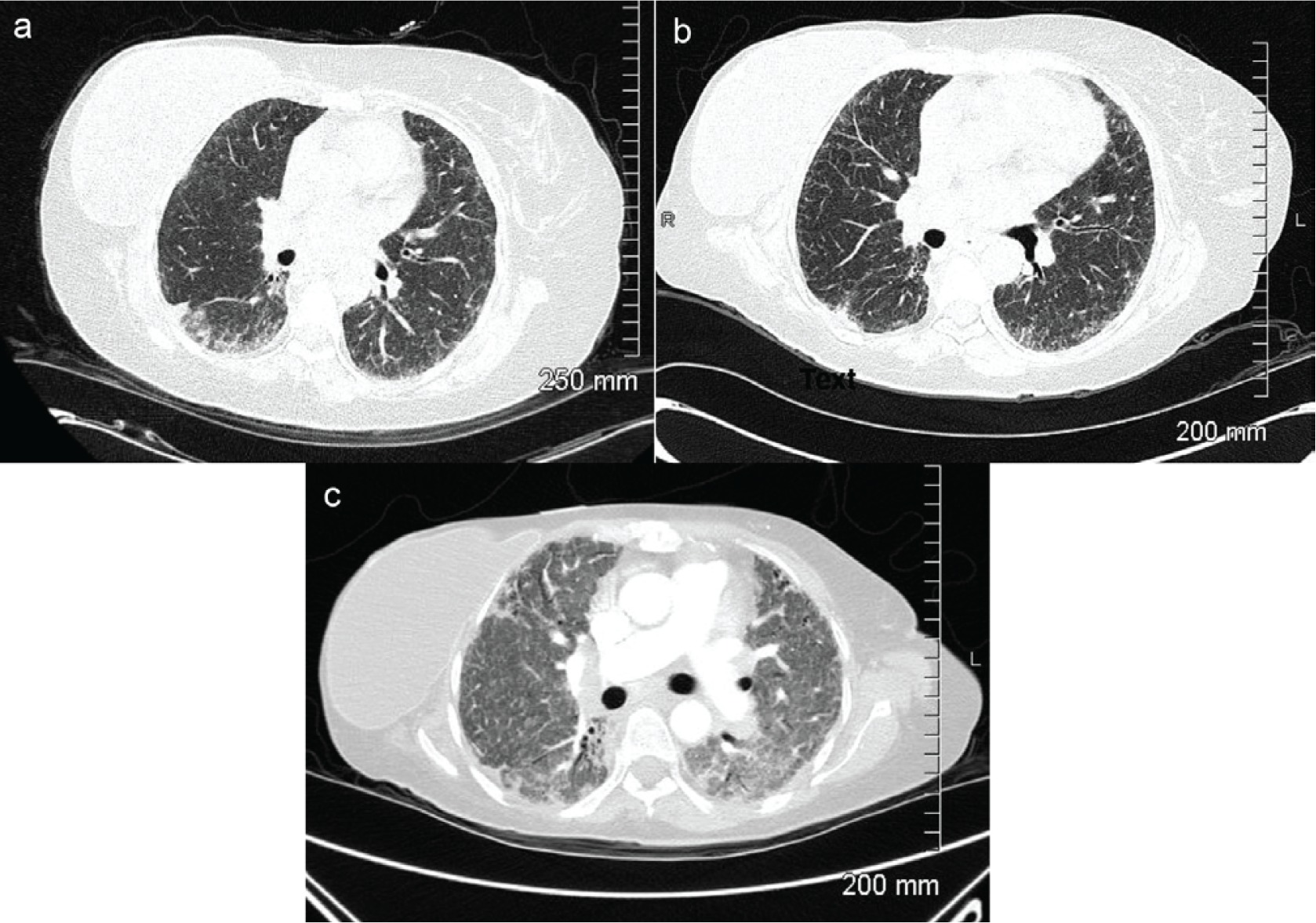
The open lung biopsy was cancelled following diagnosis of scleroderma. Increased cough was reported when pantoprazole 40 mg once a day was started. Despite therapy she continued to deteriorate with further reduced DLCO to 10%, weight loss, component of moderate pulmonary hypertension, initiation of supplemental oxygen, and hospitalization for acute exacerbation. CT images demonstrate progression of SSc-ILD (Figure 3). Around 4.5 years after the initial visit, the patient received a double lung transplant and has seen improvements in PFTs, with an increased DLCO of 45%. The most recent HRCT chest has shown stable findings and she has remained without respiratory symptoms.
Figure 3
Computed tomography (CT) scans of chest demonstrating progression of SSc-ILD with presence of significant bilateral ground class opacities; a: High resolution computed tomography (HRCT) scan at time of initial visit; b: HRCT scan 1 year 9 months after initial visit; c: CT at 4 years 2 months after initial visit during hospitalization for acute exacerbation
DISCUSSION
This case of SSc-ILD highlights the complex association between CTDs and pulmonary involvement. There is a potential of ILD in all CTDs, but the most frequent are scleroderma and rheumatoid arthritis. A CTD diagnosis can be prior or simultaneous to initial ILD findings or ILD may be the first and only manifestation of a CTD6. The uniqueness of this case is that the CTD diagnosis was made after the ILD diagnosis despite multiple initial negative autoimmune serologies. It is vital to highlight the importance of continuous evaluation for symptoms and signs of CTDs in ILD patients regardless of negative serologies.
Potential underlying CTDs are ruled out in uncharacterized ILD by serologic autoimmune tests7. In patients with established CTD, lung involvement is monitored since pulmonary complications are known to increase morbidity and mortality8. Unique to this case, is that the patient was diagnosed with ILD and had multiple negative autoimmune serologies before going on to develop scleroderma symptoms and then a positive ANA years later. In addition, there were little indications supporting a connective tissue disease diagnosis. ILD as initial manifestation in CTDs presents potential discussions about the pathogenic role of the lung as an autoimmune source9.
Establishing an accurate ILD diagnosis is important for treatment and management decisions, as these may differ between various forms of ILD such as idiopathic pulmonary fibrosis (IPF), non-IPF forms of idiopathic interstitial pneumonia, CTD-ILD, and hypersensitivity pneumonitis. Patients with suspected ILD can present with similar clinical presentations which is why evaluation is aided through patient history, physical examination, pulmonary function tests, blood tests (e.g. autoimmune serology), and thoracic imaging10. Histological patterns on chest CTs also play a role in survival and prognosis. A diagnosis of CTD-ILD has a better outcome compared to IPF partly due to the higher frequency of the nonspecific interstitial pneumonia (NSIP) pattern compared to the usual interstitial pneumonia (UIP) pattern. In addition, CTD-ILD patients with NSIP have a better prognosis than patients with idiopathic NSIP and CTD-ILD patients with UIP have a better prognosis compared to patients with IPF5.
Other manifestations of scleroderma can also include pulmonary hypertension and kidney involvement. In addition to ILD, one of the other pulmonary manifestations of scleroderma is pulmonary hypertension (PH). PH is present in approximately 30% of patients with clinically significant SSc-ILD and those with PH have a higher mortality rate than those without PH. Along with ILD, PH should also be screened for in those newly diagnosed with SSc as well as annually due to increased morbidity and mortality11.
In addition, scleroderma renal crisis (SRC) is a well-studied form of renal involvement in SSC that demonstrates significant morbidity and mortality. The exact pathogenesis of SRC is not known but believed to be associated with injury to epithelial cells. Corticosteroids have been implicated as a risk factor when used in doses greater or equal to 15 mg per day in the preceding 6 months12. Treatment with systemic oral corticosteroids is a preferred initial therapy for many ILDs, but have the potential to result in serious side effects. This further illustrates the importance of establishing a correct ILD diagnosis and continuing to explore new treatment options too that are well tolerated13.
Frequently used treatment options for SSc-ILD are immunosuppressive agents, like mycophenolate mofetil (MMF) and cyclophosphamide. MMF is an inhibitor of T and B lymphocyte proliferation and has been shown to prevent deterioration of pulmonary function in SSc-ILD. Compared to cyclophosphamide, MMF displays better long-term use and fewer adverse events. As treatment spectrum evolves, antifibrotic options approved for patients with IPF, nintedanib and pirfenidone, are likely to be important developments. Nintedanib is a tyrosine kinase inhibitor and pirfenidone is a pyridine compound that both exhibit anti-inflammatory and anti-fibrotic effects. In 2019, the SENECIS randomized, double-blind, placebo-controlled trial for nintedanib became the first pharmacological agent to secure FDA approval for SSc-ILD. Also, pirfenidone is currently being investigated in combination with MMF in Scleroderma Lung Study III (NCT03221257)14.
Limited research is known about autoantibody seroconversion for patients with SSc-ILD specifically. In a study looking at clinical features of ANA negative SSc, out of 3249 patients only 6.4% patients were ANA negative; 19 of these ANA negative patients were selected for follow-up where it was found that only 1 patient had seroconverted to ANA positive15. Gbadamssi et al.16, reported a case SSc-ILD in which the patient had exertional dyspnea for 2 years and was diagnosed with ILD; 6 months later was diagnosed with SSc upon presentation of macules on ears, edema on fingers, and digital-tip pitted scars. Swartz et al.20, also reported a case where the patient was diagnosed with SSc eight months following ILD diagnosis where there was an increase of antinuclear antibodies and presence of anticentromere antibodies.
A retrospective analysis of CTD-ILD patients during a 6-year period found that ILD was diagnosed prior to CTD in 8% of patients and simultaneously in 35% of cases. The CTD was diagnosed on average 5 months after the ILD diagnosis6. A 2017 study conducted by Alsumrain et al.7, assessed the frequency of incident connective tissue related interstitial lung disease (CTD-ILD) as detected by autoimmune serology and clinical presentation. It was found that no ILD patient with negative serology had autoimmune disease at presentation, but 2 in 605 patients (0.003%) did develop CTD-ILD, on average greater than 2 years later. It is not common for CTDs to be diagnosed following ILD diagnosis, but it is observed in a number of patients. This pattern is also seen in idiopathic inflammatory myopathy (IIM), where approximately 10% of patients were diagnosed with ILD before IIM in a 2020 retrospective study17. In a study on 2678 Chinese ILD patients, 67% were identified as having CTD-ILD. From those 1798 patients, complete clinical data were available for 1044 CTD-ILD patients. The authors found that 25% of the 1044 had negative autoantibodies at initial hospital admission which then became positive during follow-up. This illustrates the importance of examining extrapulmonary symptoms and completing serologic testing at follow-up appointments or when new symptoms arise18. ILD patients require careful clinical and immunological evaluation to ensure autoimmune causes of ILD are excluded19.
CONCLUSION
Overall, this case illustrates the importance of considering differential diagnoses during follow-up of ILD patients. Continuous monitoring of rheumatologic signs and symptoms in patients with ILD is helpful in establishing the necessary treatment and prognosis. Repeating autoimmune serology testing when there are new clinical symptoms and signs suggesting a CTD, despite previously negative results, is an area of attention illustrated by this case. This is a unique progression of SSc-ILD that demonstrates the heterogeneity and unpredictability of CTD-ILD.