


INTRODUCTION
Antiphospholipid antibody syndrome (APS) is a multisystemic autoimmune disease that is characterized by thromboembolic events and/or obstetric morbidity in the presence of antiphospholipid antibodies. The most frequent pulmonary manifestations are pulmonary thromboembolism and pulmonary arterial hypertension1,2. Recently, some rare cases have been described in which diffuse alveolar hemorrhage (DAH) appears as the initial manifestation of APS. DAH was the first presentation of APS in 9 of 79 (11%) patients3,4, and 3 out of 17 patients in one case series were diagnosed with APS only after they presented with DAH2. Yet, two other reviews, of 18 and 13 patients, noted a median DAH onset of 5.9 and 5.8 years, respectively, after the diagnosis of APS5,6.
CASE PRESENTATION
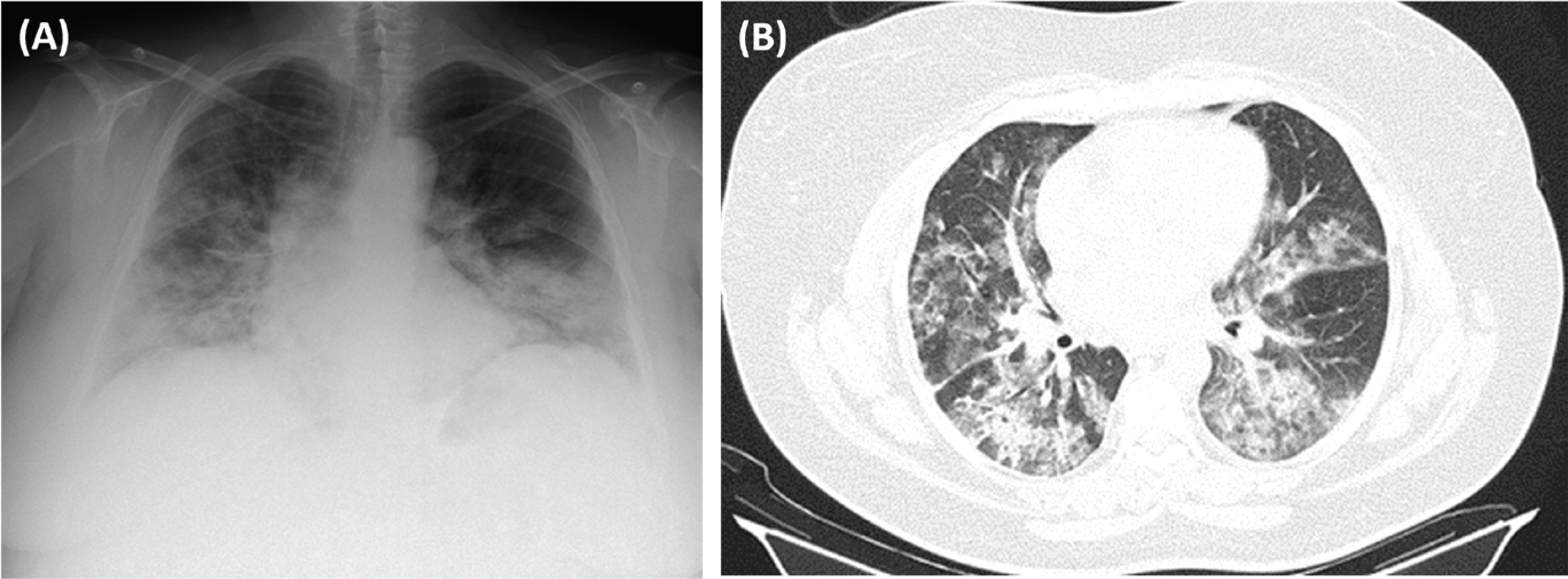
We present the case of a 44-year-old female patient, non-smoker, factory worker, with a history of sinusitis and without chronic medication, who presented to the ER with asthenia, dyspnea, and mild hemoptysis in the last month, with hemoptysis being the initial manifestation. She also mentioned photosensitive facial rash, xerostomia, and alopecia. She had no family history of thrombophilia. Her physical examination was significant for globally decreased breath sounds, a systolic heart murmur grade III/VI, and discrete edema in the lower limbs. She had no palpable lymphadenopathies. In blood gas analysis, a partial respiratory failure (Type I) was identified (pO2=58.8, pCO2=34.3, HCO3=23). Analytically, the only finding was a normochromic normocytic anemia. An echocardiography was performed, and the results were as follows: mild mitral/aortic insufficiency, and moderate aortic stenosis. A cardiologist was consulted who recommended performing an annual echocardiography. Chest X-ray (Figure 1 A) showed bilateral cotton-like infiltrates. Chest CT scan with contrast (Figure 1 B) revealed several areas of densification of the lung parenchyma, some ground glass opacities with septal thickening, particularly in the left lung field, associated with a small left pleural effusion. Pulmonary thromboembolism and active hemorrhage were excluded.
Figure 1
A) Chest X-ray (posteroanterior, PA): bilateral cotton-like infiltrates; B) Chest CT with contrast (axial plan, AP): areas of densification of the lung parenchyma, some in ground glass with septal thickening, particularly in the left lung field, associated with a small left pleural effusion
Bronchoscopy with bronchoalveolar lavage (BAL) was performed, demonstrating sequential progressive hemorrhagic fluid indicative of DAH and 80 mL of serohemorrhagic fluid were collected. Bronchial secretions were aspirated and sent for bacterial, mycobacterial, fungal stains and cultures. No microorganism was found. BAL results were as follows: neutrophils 83%, eosinophils 1%, lymphocytes 3%, macrophages 13%. She was hospitalized with a diagnosis of DAH.
In the autoimmunity study, positive-high titers of anti-cardiolipin (IgG, 233.8 UQ), anti-b2 glycoprotein (IgG 729.5 UQ) IgG antibodies and positive lupus anticoagulant were identified. There was also a slight increase in anti-dsDNA (30.3 UI/mL) and ANA (FEIA, 1.9 U/mL) antibodies. The ANCA antibodies (anti-proteinase 3, anti-myeloperoxidase) and anti-glomerular basement membrane antibodies were negative.
Complement levels, urinary sediment/urine 24 h, and Coombs test were negative. The paranasal sinus CT was normal.
Dermatology evaluation was requested. After careful evaluation, photosensitivity was excluded, and the patient’s facial rash was considered a telangiectatic rosacea. A skin biopsy was not performed as the rash rapidly improved with hydration.
DISCUSSION
The case was presented at a multidisciplinary team meeting. Based on SLE diagnostic criteria (2019), although the patient had entry criteria (ANA with a positive test - FEIA, 1.9 U/ mL), she did not present any clinical criteria or a total score ≥10 points. As a result, SLE was excluded and the diagnosis of APS with DAH as a form of presentation was established.
The patient started treatment with prednisolone (PDN) 1 mg/kg/day, mycophenolate mofetil (MMF), and hydroxychloroquine (HCQ). A few days after initiating treatment, she recovered completely and no longer needed O2 or had any additional episodes of hemoptysis.
In subsequent consultations, the doses of PDN and MMF were titrated to 0.75 mg/kg/day and 1500 mg 12/12-h, respectively. In the following 2–3 months, the patient had 2 additional hospitalizations due to recurrence of hemoptysis despite optimized therapy. In the second hospitalization, besides the episode of hemoptysis, the patient was also subfebrile and cultures to exclude infection were requested.
As she also had a fast recovery after high doses of PDN and culture results were not available, considering the risk of infection, the administration of rituximab was postponed, and she was discharged with titrated doses of PDN, MMF and HCQ. She was also immunized during this period. In the third episode, as it was considered a recurring and potential life-threatening situation, culture results were negative and the patient was already immunized, rituximab 1 g iv was associated to the therapeutic scheme.
Up to the present moment, 4 months after initiating rituximab, the patient did not present new episodes of hemoptysis.
CONCLUSION
DAH is a rare and life-threatening complication of APS and is associated with high mortality risk7. Pulmonary capillaritis, along with antibody-mediated endothelial proliferation, likely contributes to its pathogenesis. DAH typically presents with hypoxemia and subsequent respiratory failure, anemia, hemoptysis, and diffuse radiographic pulmonary infiltrates. Given the rarity of pulmonary hemorrhage in APS, treatment is guided by case reports, case series and inter-disciplinary experience. In addition to supportive care, high-dose corticosteroids are the initial treatment for DAH in APS. Corticosteroid therapy induces remission in most patients; however, recurrence may occur in up to half of cases, which requires the introduction of a corticosteroid-sparing immunosuppressant and, eventually, anti-TNF alpha, in order to maintain remission8,9.