



INTRODUCTION
Amyloid are insoluble heterogeneous proteins, with a sheet structure, that bind to the dye Congo Red and produce an apple-green birefringence under polarized light microscopy1. Amyloidosis is a rare disease characterized by abnormal extracellular amyloid deposition, which can accumulate in a wide range of organs, causing functional damage and death. Amyloidosis can be systemic or limited to a single organ. If it is pulmonary amyloidosis, it can be localized or be part of systemic amyloidosis. Tracheobronchial amyloidosis (TBA), nodular parenchymal amyloidosis and diffuse alveolar septal amyloidosis are three types of pulmonary amyloidosis2. TBA accounts for 0.5% of benign lesions in the tracheobronchial tree and can cause airway obstruction and death as the disease progresses3. We present a case of TBA in a patient with acute myeloid leukemia (AML) and hemoptysis that had previously gone unreported and was difficult to treat.
CASE PRESENTATION
A 42-year-old Libyan male patient was being followed up in the hematology service after being investigated for progressive leukocytosis (35.3×103/µL) and bicytopenia (hemoglobin, 6.4 g/dL; thrombocyte, 33×103/µL) for a month. The patient, who was diagnosed with tuberculosis two years ago as a result of recurrent hemoptysis, took anti-tuberculosis drugs for a total of six months. He has a smoking history of over 25 pack years and no history of regular drug use. Following the bone marrow biopsy to investigate leukocytosis and bicytopenia, the patient was diagnosed with AML-M3. All-trans retinoic acid (ATRA) and idarubicin chemotherapy were administered to the patient. The patient complained of increasing dyspnea, hemoptysis and cough in the weeks that followed. Physical examination revealed bilateral rhonchi and coarse crackles in the left upper zone. Because of his hemoptysis of unknown cause, pulmonary function tests could not be done on the patient who showed evidence of intrathoracic obstruction on physical examination. Thoracic CT revealed tracheal and bronchial wall thickening, sequelae in the left lung upper lobe, infected cystic bronchiectasis, and calcifications in the walls of the main and lobar bronchi. The left upper lobe bronchus and right middle lobe bronchus were obliterated, and the lung parenchyma showed atelectasis (Figures 1 and 2). Following the thorax CT findings, it was decided to perform flexible bronchoscopy for the patient’s endobronchial evaluation. Flexible bronchoscopy revealed diffuse submucosal hypertrophy extending from the trachea to the main bronchi, as well as fragile and bleeding-prone necrotic and anthracotic lesions narrowing the trachea and bronchial lumen (Figure 3). Punch biopsies were taken from submucosal hypertrophy areas narrowing the left main bronchus. Bronchial lavages were taken from the left lung upper lobe bronchus and the right lung middle lobe bronchus. In the patient with a history of tuberculosis, endobronchial tuberculosis was considered, but Ehrlich-Ziehl-Neelsen (EZN) stain and PCR for M. Tuberculosis were negative in bronchial lavage. Pseudomonas aeruginosa was detected from BAL (>105 CFU/mL) cultures. Amyloid deposition in the Lamina Propria was observed in the pathology of the bronchoscopic punch biopsy, and Congo Red staining was positive in the histochemical examination (Figure 4). As a result of the thorax CT, bronchoscopy images and pathology report, the patient was diagnosed with TBA. Due to the fact that the patient had a hematological disease such as AML, it was thought that TBA may be associated with systemic amyloidosis, albeit rare and the patient was investigated for systemic amyloidosis. Serum amyloid A protein (SAA) and serum protein electrophoresis were normal and there was no proteinuria in the urine. The patient’s severe hemoptysis persisted after the flexible bronchoscopy. Despite fresh frozen plasma, thrombocyte and erythrocyte replacements, the patient’s AML-related thrombocytopenia and anemia worsened. Although TBA necessitated rigid bronchoscopy and stent insertion, the procedure was deemed high risk due to the patient’s poor general condition and complications from AML. Despite the start of combined antibiotic therapy for carbapenem-resistant pseudomonas (cefepime, levofloxacin, teicoplanin, fosfomycin), the increase in infectious markers persisted and the patient was followed up in the ICU (Intensive Care Unit). As the patient’s tachypnea and respiratory acidosis worsened while on noninvasive mechanical ventilation (NIMV) in the ICU, he was intubated and connected to a mechanical ventilator. One day after, the infection markers increased (Procalcitonin: 10.4 ng/mL, CRP: 336 mg/L) and despite combined antibiotherapy and fluid replacement therapy, the patient died from sepsis.
Figure 1
(A-H) Axial thorax CT images show sequelae changes in the left upper lobe of the lung, widespread thickening of the trachea and bronchial walls, obliteration of the left upper lobe bronchus and right middle lobe bronchus, cylindrical and cystic bronchiectasis, more prominently in the left upper lobe, and atelectasis in the right middle lobe
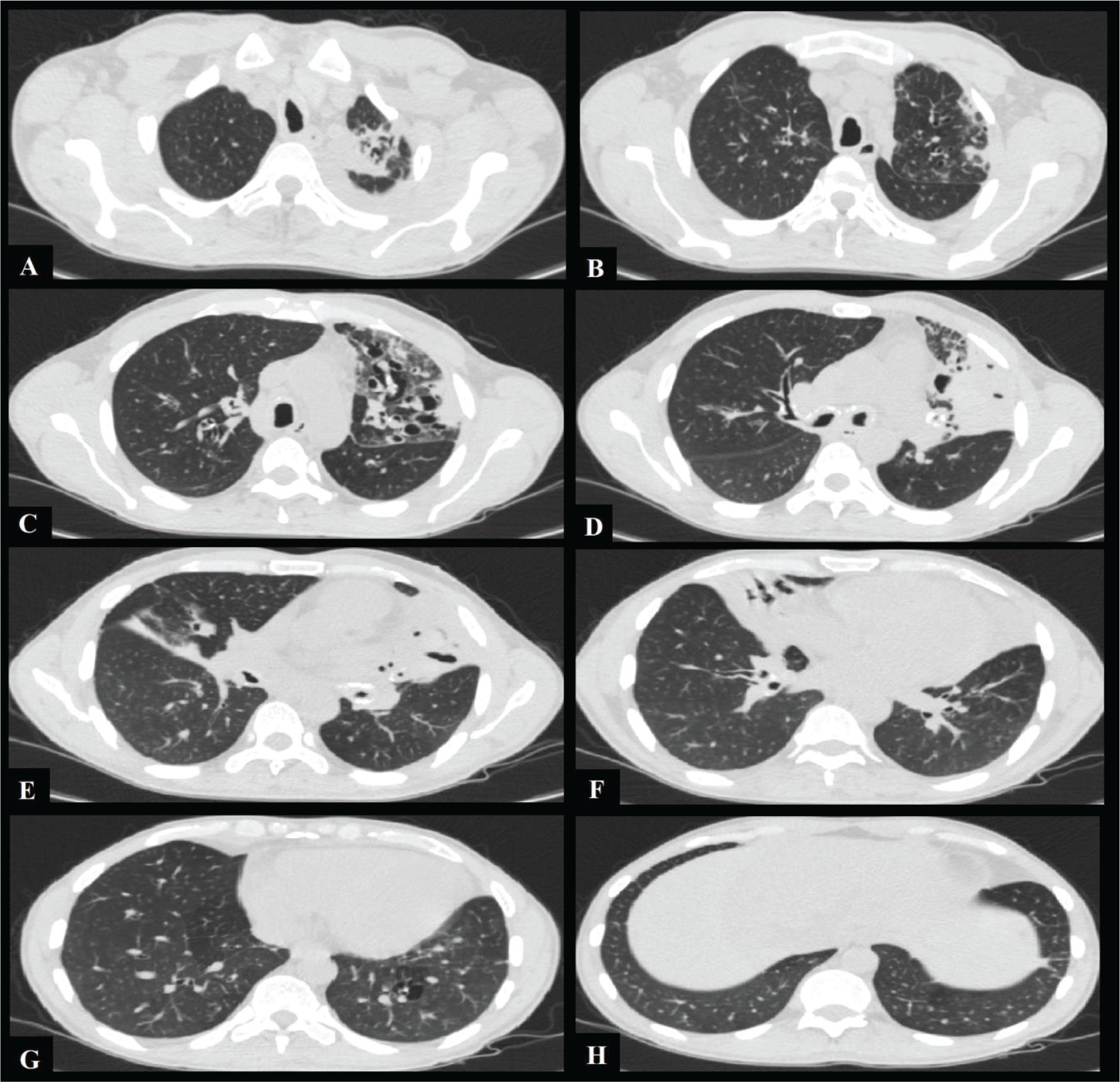
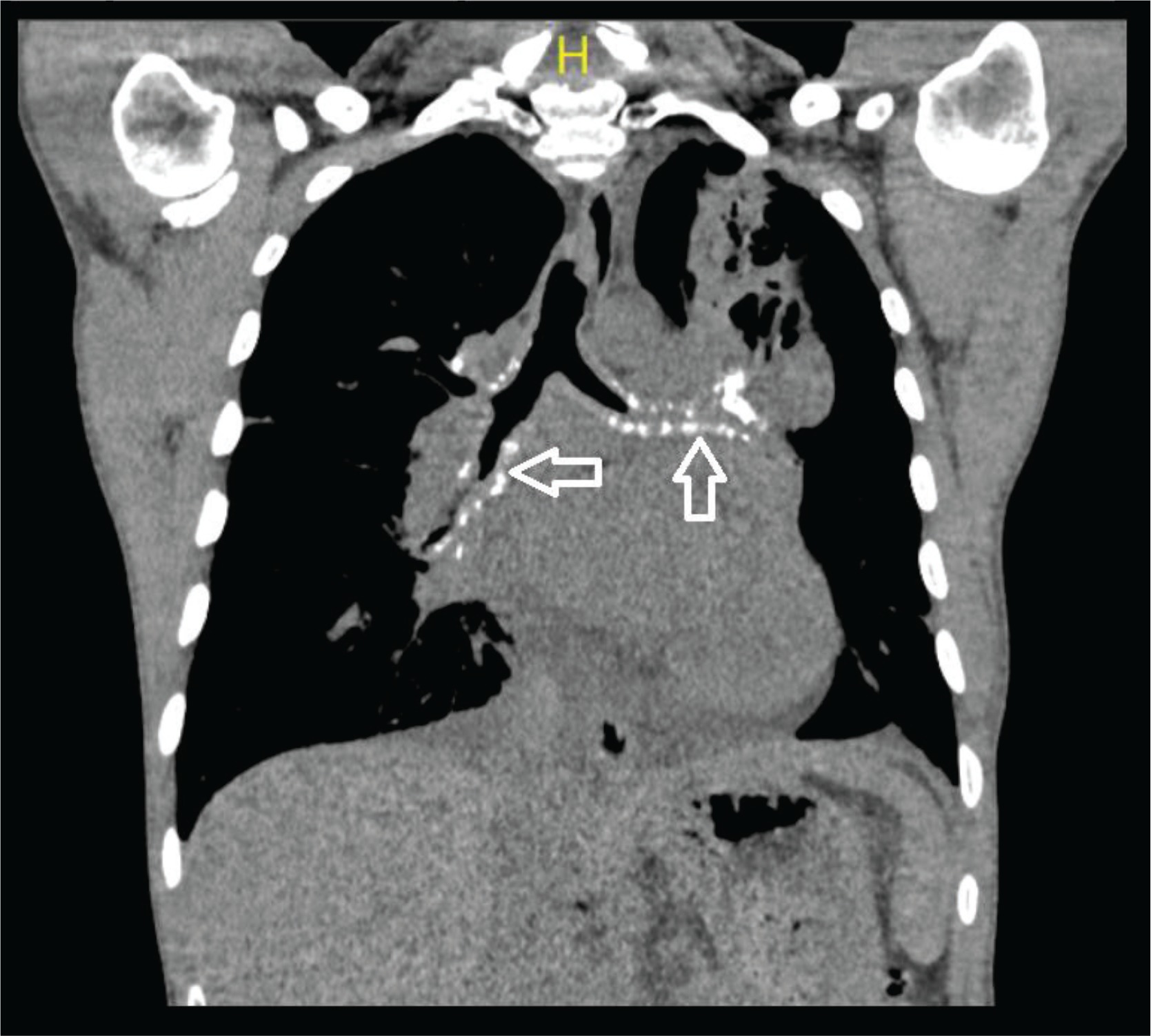
Figure 2
Calcified amyloid deposition seen under the tracheal bronchial mucosa in the coronal section of the mediastinal window in thorax CT (white arrow)
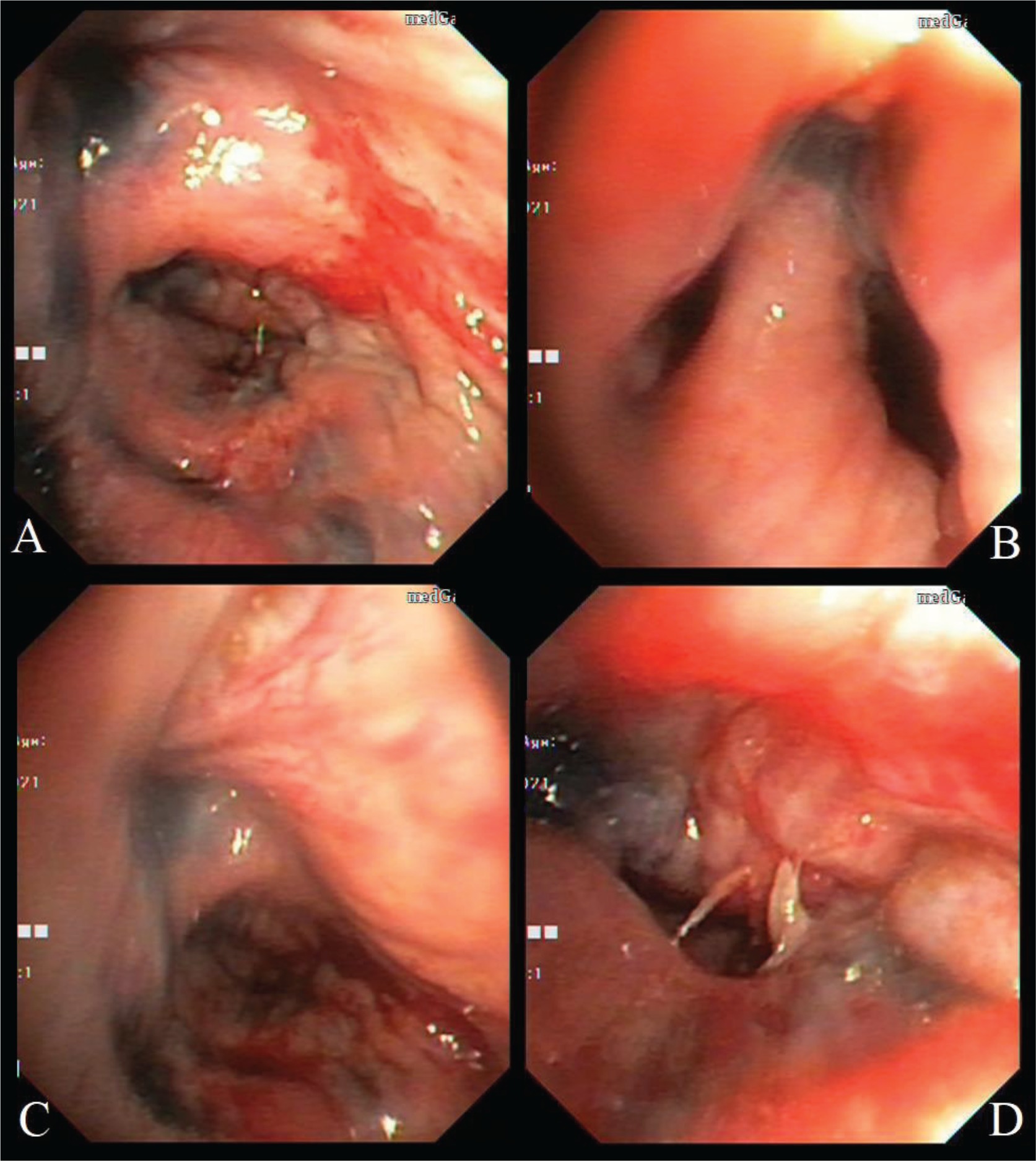
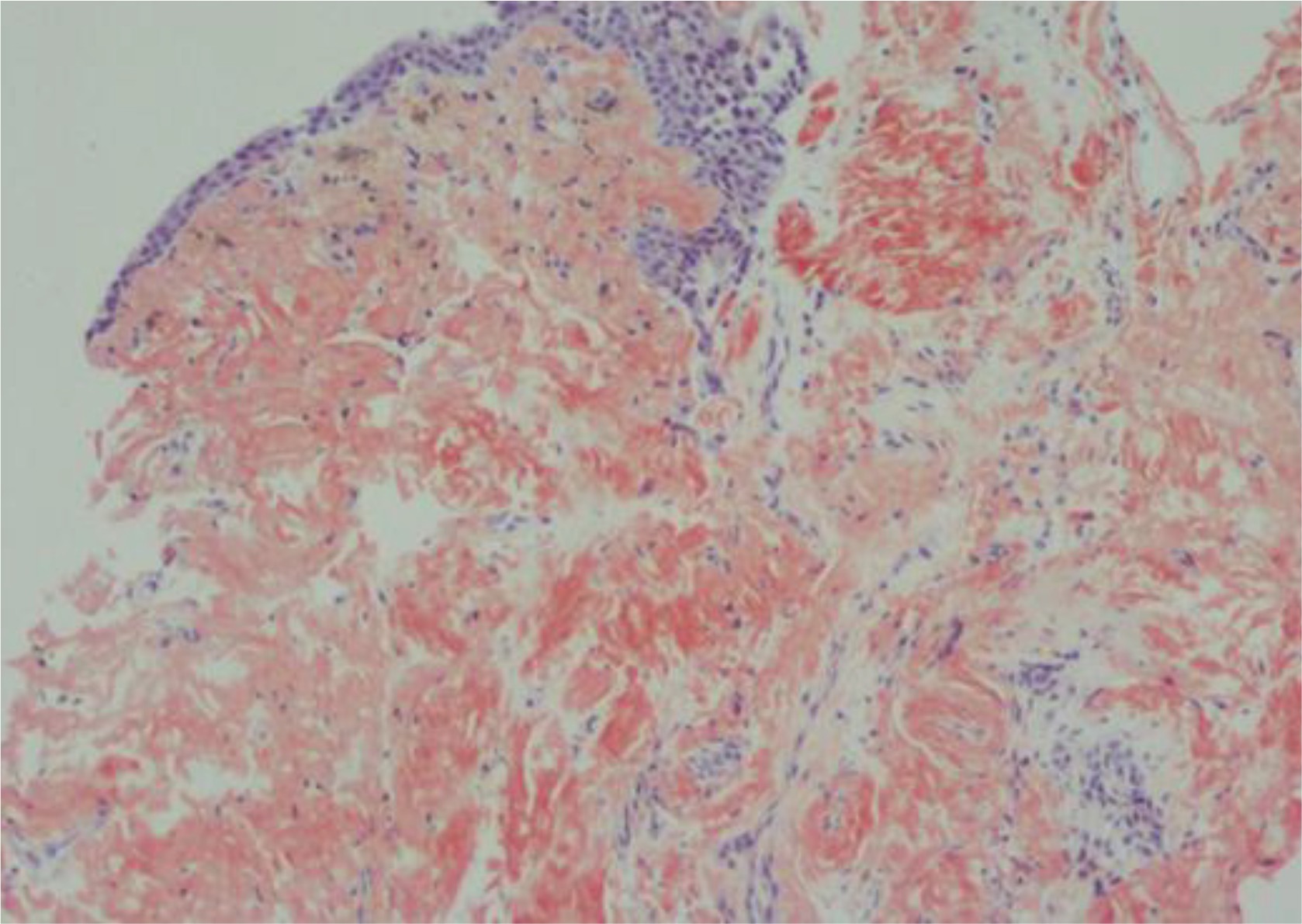
DISCUSSION
The incidence of amyloidosis is approximately 1.2 per 100000 individuals per year worldwide. Systemic amyloidosis can be seen as primary and secondary amyloidosis. Primary amyloidosis is caused by the deposition of immunoglobulin lambda light chain (AL protein) as a result of cell dyscrasias; secondary amyloidosis, is caused by the accumulation of serum amyloid A (SAA) of hepatic origin as a result of chronic inflammatory/autoimmune diseases such as bronchiectasis, tuberculosis, sarcoidosis, cystic fibrosis, collagen vascular diseases4.
TBA is most commonly associated with local AL amyloidosis. Tracheobronchial amyloidosis (TBA) accounts for about 1.1% of referrals to an amyloid center5. Tracheobronchial amyloidosis is a condition that primarily affects men in their fifth or sixth decade. TBA is characterized by localized, diffuse or multifocal accumulation of amyloid material as submucosal plaques and/or polypoid tumorlike mass. It can appear as plaques narrowing the entire tracheobronchial tree or its major parts, and it can occur as isolated endobronchial or tracheal tumors. The amount of amyloid deposited in the airways is directly related to morbidity and mortality. The cause of death is progressive bronchial obstruction and respiratory failure. Although the disease can be completely asymptomatic, symptoms such as dyspnea, wheezing, hemoptysis, recurrent pneumonia, cough, and atelectasis can occur2,4.
The diagnosis is made using a thoracic CT, bronchoscopy, and a biopsy. Endobronchial disease and systemic disease are usually unrelated. As a result, requesting costly further investigations for evidence of systemic amyloidosis is often not advised. Although TBA is mostly localized and unrelated to systemic amyloidosis, when we look at the literature, Capizzi et al.6 investigated 14 patients out of 17 TBA cases for systemic amyloidosis and identified one patient with a previous diagnosis of multiple myeloma. Chest radiographs are normal in half of the patients, but others may have atelectasis or vascular collapse, calcifications, bronchiectasis, or hilar lymphadenopathies4,5.
It is difficult to distinguish between localized tracheobronchial amyloidosis and endobronchial malignancy on bronchoscopic examination. Submucosal lesions and pale plaques, nodules or polyps on bronchoscopy may indicate areas of localized bronchostenosis. During bronchoscopy, submucosal bleeding foci caused by lesions may be seen. On thoracic tomography, significant thickening and narrowing of the tracheal, main bronchi and segmental bronchi can be seen. Tracheopathia osteoplastica, calcification, and ossification of subcutaneous nodules along the tracheobronchial tree can be seen in diffuse TBA7,8. Histopathological examination of bronchial biopsies demonstrating amyloid accumulation and Congo Red positivity leads to a definitive diagnosis9.
Rigid bronchoscopy with endobronchial debulking, cryotherapy, stent placement, colchicine use, and external beam radiation are all treatment options. External beam radiation appears to be effective at reducing local plasma cell proliferation, vasculature, inflammatory cell migration, and other aspects of the local environment surrounding amyloid deposits. Radiation-induced esophagitis and pneumonitis are two possible side effects. Other modalities have limitations such as lesion access, bleeding, and a higher recurrence risk2. Pharmacological interventions, such as corticosteroids and colchicine, may be beneficial in systemic amyloidosis, but should not be used as the primary treatment for localized TBA8,10.
Because TBA causes airway obstruction, patients may have been misdiagnosed with asthma and COPD for many years and may have received insufficient or wrong therapy. Clinicians must make an early diagnosis of TBA in patients before symptoms of airway obstruction progress and lead to secondary pulmonary problems. In situations of TBA discovered early, invasive bronchoscopic procedures and radiation treatments may alleviate symptoms and extend patients quality of life and life expectancy.
The reported prognosis may vary according to the studies performed. One study reported 70% survival after 7 to 12 years, while others reported 31% and 43% after 4 to 6 years. The 5-year survival rate for diffuse submucosal transbronchial amyloidosis has been reported to be 30% to 50%. The location and extent of airway involvement, as well as the presence of systemic disease, are likely to influence prognosis2,8,11.
There are a few rare case reports of AML accompanied by systemic amyloidosis in the literature. A familial Mediterranean fever patient with a history of AML was diagnosed with systemic amyloidosis after developing renal failure12. In another case report, the association of Multiple Myeloma, AML and light chain amyloidosis was reported13.
So far, no literature has defined the coexistence of AML and localized TBA. The appearance and increased severity of pulmonary symptoms related to TBA following the diagnosis of AML in our patient make us think that the two entities are related. Extensive research is required to explain the association of myeloproliferative diseases and amyloidosis. As in our case, the poor course of AML-related disease, recurrent resistant infections and bleedings due to TBA may complicate the management and treatment of the disease.
CONCLUSION
Tracheobronchial amyloidosis (TBA) is a rare disease that can be easily pathologically diagnosed using flexible bronchoscopy. Thorax CT findings in asymptomatic patients may aid in the early diagnosis. With the endobronchial spread and progression of the disease, dyspnea, hemoptysis and recurrent infections may occur. The coexistence of TBA and AML has never been reported in the literature. As in our case, the management of complications related to the primary disease can be difficult and it should not be forgotten that some cases can be fatal.